Stefan Linquist
Hi Larry,
First, thank you for giving my paper a careful read. The intended audience is the community of biologically-minded philosophers who seem largely convinced that:
1) Genes are so passé. More specifically, when it comes to explaining phenotypic development and evolution, such non-genic factors as noncoding RNA, maternally inherited methylation patterns, repetitive elements, etc. are equally if not more significant than genes. It is a short step to the view that most of these elements are somehow functional for the organism. Stated pejoratively, thinkers like John Mattick and Evelyn Fox-Keller have had a significant intellectual founder-effect on my discipline. My paper attempts to push back against this trend.
2) Molecular biology can and should ignore evolution. The idea here is that when it comes to the search for molecular mechanisms, it doesn’t matter if genomes are the product of multi-level evolution or if they had been created by God. When you work on mechanisms, you do experiments, and evolutionary considerations are irrelevant to how those experiments are conducted and interpreted. Or, so the thinking goes.
Many of your blog posts present counter arguments to these ideas with a level of understanding and precision that exceeds my efforts in this paper. Nonetheless, I want to take issue with your one suggestion (if I understand correctly) that biochemists tend to operate with a sophisticated understanding of the genome. My position is that biochemistry might be necessary, but is not sufficient for an informed view of genomics. Without Darwinian reasoning, biochemistry leads down unnecessary blind alleys.
Obvious to whom?
Let me be upfront that I am something of an academic bumpkin in comparison to fancy city-folk like you, or Palazzo, or Graur, or Doolittle, or Haig. My knowledge of molecular genetics is largely self taught. This is partly why I am perplexed by statements like the following. In a special collection of Chromosome Research entitled, “Transposable elements and the multidimensional genome” (2018), P.A. Larson (the collection editor) opens with this doozy:
“There is no such thing as “junk DNA.” Indeed, a suite of discoveries made over the past few decades have put to rest this misnomer and have identified many important roles that so-called junk DNA provides to both genome structure and function…”
Is it me? Or is it him? It’s him, right? My point is simply that it can’t be obvious to everyone within the molecular biological community that not every binding site or repetitive element is somehow functional for the organism. This is to say nothing of the hype surrounding lncRNA.
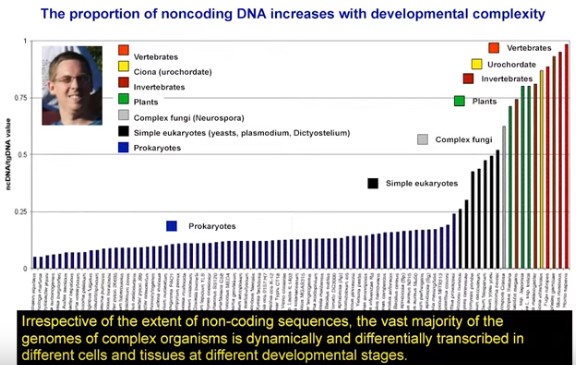
My argument in the paper is that the missing piece of information is an understanding of where the majority of eukaryotic DNA comes from: a byproduct of coevolutionary interactions between parasitic TEs and the cell. Indeed, I provide evidence in another publication, Transposon dynamics and the epigenetic switch hypothesis, that over the past two decades or so, within the fields of molecular biology and biomedicine, interest in TEs has steadily declined. This trend is surprising given that over the same period we have come to learn just how prevalent TEs are in most genomes. I think that I can show, in another forthcoming paper, that this trend toward ignoring TE coevolutionary dynamics is associated with the increased biomedicalization of molecular biology as a discipline (more on that another time, perhaps). Whether this decline of interest in TEs is responsible for the tendency to interpret junk DNA as somehow functional is a further question.
Another factor that I find perplexing is the trickle of molecular biology majors who attend my philosophy of biology undergraduate seminar. I'm not surprised that they show up at all, rather I'm surprised about their conviction that any biochemically active region of the genome simply must be functional for the organism. "Functional until proven otherwise" seems to be the mantra that one must memorize in order to pass the med-school admissions exam. When I suggest to them that Darwinian reasoning leads to an alternative hypothesis about most of the DNA in eukaryotic genomes, they balk. Some just leave my class: “What does he know, just a philosopher.” Such is the life of an academic bumpkin from the intellectual sticks.
This is all to say that, yes, you are correct that my paper presents no new biological data. In a sense, it is old news. But it is news that many people –even some academic city slickers-- seem not to have absorbed.
I like to think of philosophy journals as a clearing house for discussions that are extremely important, but would be unlikely to elbow their way into the pages of most scientific journals. Aside of helpful blogs such as yours, where else are we to debate the theoretical framing and interpretation of junk DNA?
What’s with the philosophical obsession over functions?
It’s true that my article focuses on this longstanding debate over CR vs SE functions. I can imagine that from the perspective of a molecular biologist (with such a rich ontology to draw from, and so many fine grained distinctions at your disposal) this binary must appear ham-fisted.
Let me say two things. First, I repeat that my main audience is the community of biological philosophers. In this context, these basic categories of function and the debates that surround them provide a lingua franca. To have this discussion without connecting it to function concepts would seem odd. Second, I think that you and I would both be happy if the word “function” in genetics were restricted to what I elsewhere call maintenance functions. That is, to elements that have been maintained by purifying selection. However, many of my colleagues are so convinced of point 2 (above) that this proposal is essentially a non-starter. That is, they maintain that since molecular biology doesn't investigate causal role functions (a big assumption, but let it go for now), then this discipline can ignore Darwinian reasoning. My argument is that this inference is too quick. A problem with CR functions is their permissiveness: any old strand of DNA can have some CR function or other. What we need is some way to sort the functional wheat from the junky chaff. To do that, thinking about selective history is your best bet. In effect, you can deny entrance to Darwin at the front door if you want, but eventually you’ll have to let him in through the back.
A final note on the term “selective history.” You suggested that I should have instead used “evolution” in order to discourage a Panglossian view of the genome. The issue I see with your suggestion is that “evolution” is too vague –it really just means change over time. My contention is that one needs to do more than just consider historical (e.g. phylogenetic) details in order to take a biologically informed view of the genome. In addition, one needs to think about how the cell coevolves with parasitic TEs. Maybe “coevolutionary dynamics” would have been a better choice.
Finally, a plug. The paper you read is part of a special collection in Biology and Philosophy that I co-edited with Ford Doolittle entitled, “Function, junk and transposable elements: contested issues in the science of genomics.” As I write, I see that three papers have so far appeared and the other two (including mine and one coauthored by Alex Palazzo) should see the light of day soon:
Hopefully some of these will provide additional fodder.Function, junk and transposable elements: contested issues in the science of genomics
Cheers,
Stefan