
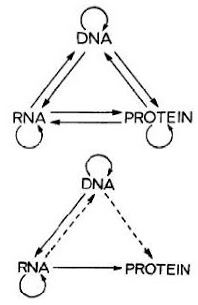
When doing an exeriment, it's important to keep the number of variables to a minimum and it's important to have scientific controls. There are two types of controls. A negative control covers the possibility that you will get a signal by chance; for example, if you are testing an enzyme to see whether it degrades sugar then the negative control will be a tube with no enzyme. Some of the sugar may degrade spontaneoulsy and you need to know this. A positive control is when you deliberately add something that you know will give a positive result; for example, if you are doing a test to see if your sample contains protein then you want to add an extra sample that contains a known amount of protein to make sure all your reagents are working.
Lots of controls are more complicated than the examples I gave but the principle is important. It's true that some experiments don't appear to need the appropriate controls but that may be an illusion. The controls might still be necessary in order to properly interpret the results but they're not done because they are very difficult. This is often true of genomics experiments.































{kind=link}
{kind=link}