Here's a link to a recent review on the origins of SARS-CoV-2 written by 21 scientists from seven different countries. They are all recognized experts on the subject.
Holmes, E.C., Goldstein, S.A., Rasmussen, A.L., Robertson, D.L., Crits-Christoph, A., Wertheim, J.O., Anthony, S.J., Barclay, W.S., Boni, M.F., Doherty, P.C., Farrar, J., Geoghegan, J.L., Jiang, X., Leibowitz, J.L., Neil, S.J.D., Skern, T., Weiss, S., R, Worobey, M., Anderson, K.G., Garry, R.F. and Rambaut, A. (2021) The origins of SARS-CoV-2: A critical review. Cell. (published online Aug. 19, 2021) [doi: 10.1016/j.cell.2021.08.017]
Since the first reports of a novel severe acute respiratory syndrome (SARS)-like coronavirus in December 2019 in Wuhan, China, there has been intense interest in understanding how severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged in the human population. Recent debate has coalesced around two competing ideas: a “laboratory escape” scenario and zoonotic emergence. Here, we critically review the current scientific evidence that may help clarify the origin of SARS-CoV-2.
If you are interested in the origin of this virus then you must read this paper, especially if you are a supporter of the lab leak conspiracy theory. The review attempts to put the issues into the proper perspective by looking at the big picture instead of focusing on the kinds of trivia promoted by conspiracy theorists. For example, the review notes that SARS-CoV-2 is the ninth documented coronavirus that has spread to humans and all of the previous ones have zoonotic origins as do the vast majority of human viruses. Four of these viruses are endemic (HCoV-OC43, HCoV-HkU1, HCoV-229E, and HCoV-NL63). They cause minor respiratory illnesses that we classify as common colds. Its interesting to learn about HCoV-HKU1 ...
"These [endogenous] viruses have zoonotic origins, and the circumstances of their emergence are unclear. In direct parallel to SARS-CoV-2, HCoV-HKU1, which was first described in a large Chinese city (Shenzhen, Guangdong) in the winter of 2004, has an unknown animal origin, contains a furin cleavage site in its spike protein and was originally identified in a case of human pneumonia (Woo et al., 2005)."
The authors look at the bat coronaviruses that have been isolated and sequenced before and after the pandemic began. In particular, they examine RaTG13, a bat virus that was identified at the Wuhan Institute of Virology (WIV). They note that this virus is not particularly closely related to SARS-CoV-2 and that three other bat viruses that were identified only last year are more closely related to SARS-CoV-2. The sequences of all these viruses were examined because there has been unfounded speculation that scientists at the WIV created SARS-CoV-2 from RaTG13.
"Collectively, these data demonstrate beyond reasonable doubt that RaTG13 is not the progenitor of SARS-CoV-2, with or without laboratory manipulation or experimental mutagenesis."
Furthermore, the authors note that the RaTG13 virus didn't actually exist. It was just a bunch of sequences assembled in silico from short sequence reads.
The lab leak conspiracy theorists imagine that the WIV was working on SARS-CoV-2 prior to the beginning of the pandemic and they somehow allowed to virus to escape and infect the citizens of Wuhan. The experts examined this speculation and conclude ...
"... there is no data to suggest that the WIV—or any other laboratory—was working on SARS-CoV-2, or any virus close enough to be the progenitor, prior to the COVID-19 pandemic."
This is a very important point. There is no evidence to support the most important part of the lab leak claim. It's just unfounded speculation.
What about the claim that some workers at the WIV were hospitalized with COVID-19 in November 2019? This claim was spread on Reddit and made it's way into some US intelligence briefings in Januray 2020.
"Despite extensive contact tracing of early cases during the COVID-19 pandemic, there have been no reported cases related to any laboratory staff at the WIV, and all staff in the laboratory of Dr. Shi Zhengli were said to be seronegative for SARS-CoV-2 when tested in March 2020 (World Health Organization, 2021), with the laboratory reportedly following the appropriate biosafety protocols during their coronavirus work (Cohen, 2020)."
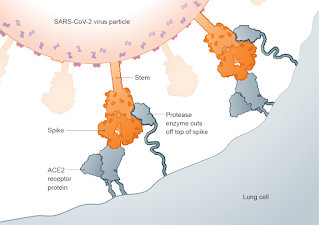
This is another important point because if it were true that workers had been infected with SARS-CoV-2 then it would support the lab leak theory. But the scientists at the WIV deny that any such infections occurred and reported to WHO that nobody tested positive for the virus. Furthermore, the public version of the American review of intelligence reports on the origin of SARS-CoV-2 published last month failed to mention the speculation that WIV workers were infected [Intelligence Community: Unclassified Report]. The most logical explanation is that the intelligence agencies could not back up the claim. If they had evidence to support the Reddit rumour then you can bet they would have made it public.
What this means is that, in the absence of evidence, the lab leak supporters have to add an additional layer to their accusation. They have to assume that the scientists at the WIV are lying and covering up the fact that they were secretly working with SARS-CoV-2 and allowed it to escape. The speculation becomes a conspiracy theory.
There's another aspect to this false claim that's very interesting. The 21 experts note that in order for there to have been multiple hospitalizations of lab workers in November 2019 there would have to have been widespread circulation of the virus since not all infections result in hospitalizations. However, a close examination of the known cases coupled with extensive modeling indicates that the virus probably started with a single individual around November 1st.
Epidemiological modeling suggests that the number of hypothetical cases needed to result in multiple hospitalized COVID-19 patients prior to December 2019 is incompatible with observed clinical, genomic, and epidemiological data (Pekar et al., 2021).
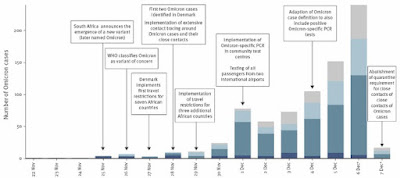
The conspiracy fans will no doubt claim that this is just part of a massive coverup by the Chinese government. This coverup involves hundreds of patients who were sick with COVID-19 in October and November 2019 and many who were hospitalized. They have all been silenced along with dozens and dozens of health care workers. Not only that, the Chinese government has been clever enough to release just enough information to make it look like the pandemic stated with a few individuals in November and spread slowly until late December as shown in the figure accompanying the review paper.
The available evidence doesn't rule out a lab leak but it's inconsistent with the Reddit rumour that three or four WIV workers were hospitalized with COVID-19 in November.
Nobody can definatively rule out a conspiracy theory—that's why they are so popular. But you can address false claims that presumably support the conspiracy theory. That's why the experts considered whether the scientists at WIV had been working on gain-of-function research or had previously worked with infectious human viruses. They could find no such evidence.
"There is no rational experimental reason why a new genetic system would be developed using an unknown and unpublished virus, with no evidence nor mention of a SARS-CoV-2-like virus in any prior publication or study from the WIV (Ge et al., 2012; Hu et al., 2017; Menachery et al., 2015), no evidence that the WIV sequenced a virus that is closer to SARS-CoV-2 than RaTG13, and no reason to hide research on a SARS-CoV-2-like virus prior to the COVID-19 pandemic. Under any laboratory escape scenario, SARS-CoV-2 would have to have been present in a laboratory prior to the pandemic, yet no evidence exists to support such a notion and no sequence has been identified that could have served as a precursor."
Sandwalk readers will recognize the issue here. You can't prove a negative so the lab leak conspiracy theorists will always be able to say that their conspiracy theory hasn't been disproven. If that's all you have to go on then I feel sorry for you. You are in the same group as those who believe in all other conspiracy theories.
The 21 experts then go on to address the claim that the virus was "pre-adapted" to humans suggesting that scientists at the WIV had selected the virus for its ability to infect human cells. This speculation includes the ridiculous claim that the furin cleavage site had been deliberately engineered. That particular claim has been debunked but so has the general claim of pre-adaption. In fact, the data shows that the virus was a generalist virus that has gradually become more adapted to humans during the pandemic. This fact, that it was a generalist virus, has been known for more than a year but that hasn't squelched the conspiracy theorists.
"SARS-CoV-2 is also notable for being a host generalist virus (Conceicao et al., 2020), capable of efficient transmission in multiple mammalian species (including mink, tigers, cats, gorillas, dogs, raccoon dogs, and ferrets), and large outbreaks have been documented in mink with spill-back to humans (Oude Munnink et al., 2021) and to other animals (van Aart et al., 2021). Combined, these findings show that no specific human “pre” adaptation was required for the emergence or early spread of SARS-CoV-2, and the claim that the virus was already highly adapted to the human host (Zhan et al., 2020), or somehow optimized for binding to human ACE2, is without validity."
The authors conclude that the lab leak conspiracy theory has no merit because there's no evidence to support it. Thus, the only reasonable hypothesis is a natural origin and there's plenty of evidence to support that hypothesis.
"There is currently no evidence that SARS-CoV-2 has a laboratory origin. There is no evidence that any early cases had any connection to the WIV, in contrast to the clear epidemiological links to animal markets in Wuhan, nor evidence that the WIV possessed or worked on a progenitor of SARS-CoV-2 prior to the pandemic. The suspicion that SARS-CoV-2 might have a laboratory origin stems from the coincidence that it was first detected in a city that houses a major virological laboratory that studies coronaviruses. Wuhan is the largest city in central China with multiple animal markets and is a major hub for travel and commerce, well connected to other areas both within China and internationally. The link to Wuhan therefore more likely reflects the fact that pathogens often require heavily populated areas to become established (Pekar et al., 2021).
We contend that although the animal reservoir for SARS-CoV-2 has not been identified and the key species may not have been tested, in contrast to other scenarios there is substantial body of scientific evidence supporting a zoonotic origin. Although the possibility of a laboratory accident cannot be entirely dismissed, and may be near impossible to falsify, this conduit for emergence is highly unlikely relative to the numerous and repeated human-animal contacts that occur routinely in the wildlife trade. Failure to comprehensively investigate the zoonotic origin through collaborative and carefully coordinated studies would leave the world vulnerable to future pandemics arising from the same human activities that have repeatedly put us on a collision course with novel viruses."
Image credits: The first figure is from the Holmes et al. paper and the second one is from a Scientific American article [A Visual Guide to the SARS-CoV-2 Coronavirus]