Take any vaccine you can get whenever you can. Moderna is the probably the very best vaccine and Pfizer-BioNTech is a close second. AstraZenica is very good but Johnson & Johnson not so much.
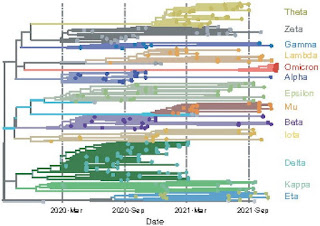
A brief summary of the COVID-19 vaccines was published in the Dec. 23rd issue of Nature. It doesn't go into a lot of details but I think the overall impressions are valid. The most serious probem with the summary is that it doesn't take into account the Omicron variant.
Mallapaty, S., Callaway, E., Kozlov, M., Ledford, H., Pickrell, J. and Van Noorden, R. (2021) How COVID vaccines shaped 2021 in eight powerful charts. Nature 600:580-583. [PDF] The extraordinary vaccination of more than four billion people, and the lack of access for many others, were major forces this year — while Omicron’s arrival complicated things further.
The first graph shows the popularity of the major vaccines. It's significant for two reasons. First, people in North America don't realize that the AstraZenica vaccine has made such an enormous contribution to fighting the pandemic. That's because AstraZenica wasn't approved in the United states in spite of its effectiveness and it got a bad reputation in Canada.
Second, the Chinese vaccine, CoronaVac (also known as Sinovac), has been widely distributed throughout the world. The CoronaVac vaccine is an inactivated virus vaccine that doesn't require ultracold temperatures for storage and it is relatively cheap to manufacture. China has been vaccinating people everywhere, notably in Brazil and Indonesia. The CoronaVac vaccine was quite effective against the early variants but it doesn't work as well with the Omicron variant.
The distribution data also shows that the Pfizer-BioNTech vaccine, the one developed in Germany, is far more popular than the Moderna vaccine that was developed in the United States. Even Sinopharm, another Chinese vaccine, is more popular than Moderna. As far as most of the world is concerned, it's the German, British, and Chinese vaccines that are going to save them and not the one created in Boston.
Some of the vaccines are more effective than others but unfortunately the Nature article only addresses the vaccines that are widely used in Europe and North America. The data shows that the mRNA vaccines are very effective against all of the variants that arose before Omicron. The mRNA vaccines not only protected against symptoms but also against severe disease (hospitalizations). The AstraZenica vaccine was also very good but not quite as good as the mRNA vaccines. The Johnson & Johnson vaccine was much less effective.
These data do not address any possible side effects of these vaccines and that's important because it is widely believed in some countries that the AstrZenica vaccine poses a much higher risk of side effects. That's not true. There may be a slightly increased risk of side effects with AstraZenica but it's not significant.
The vaccine's ability to block symptoms depends on the antibody levels in the serum while the ability to prevent long-term infections depends on the development of robust memory B-cells and T-cells. As with all vaccines, the initial antibody levels fall after the vaccination so the ability to prevent initial infections by the virus wanes over time [The omicron variant evades vaccine immunity but boosters help] [On the effectiveness of vaccines].
You can see from the above graph that the vaccines' ability to prevent infecion by the Delta variant falls off considerably by six months after completing the vaccination schedule. It's important to note that this data is with the Delta variant and it explains why countries that rushed to vaccinate their population as quickly as possible in early 2021 suffered more in the Delta wave. It's why booster shots were promoted in Israel and the United States because both of those countries vaccinated early and waited only the minimal time between doses. (Other countries waited longer between the first and second doses so the waning of initial infection was delayed.)
The waning effect is even more pronounced with the Omicron variant because it arose later in the year when far more people were beyond the six month limit of primary infection protection. What this means, I think, is that the Omicron variant isn't special because it "escapes immunity"—that would have been true of any new variant just as it was true of Delta. In any case, the mRNA vaccines are better because they start with a higher level of protection and if the data is accurate it means that Moderna is better than Pfizer.
I was prompted to post this article because many Canadians are hesitant to get the Moderna vaccine for their booster shot, especially if they had Pfizer first. That's ridiculous. Moderna is probably a bit better and, besides, there's plenty of data showing that mixing vaccines is better than sticking with the same one for all your shots.
Image Credit: The coronavirus figure is from Alexy Solodovnikov and Wikmedia Commons.