
This is my fifth post on the complete telomere-to-telomere sequence of the human genome in cell line CHM13 (T2T-CHM13). There were six papers in the April 1st edition of Science. My posts on all six papers are listed at the bottom of this post.
This is my fifth post on the complete telomere-to-telomere sequence of the human genome in cell line CHM13 (T2T-CHM13). There were six papers in the April 1st edition of Science. My posts on all six papers are listed at the bottom of this post.
How can different molecular biologists have such opposite views of the history of their field?
I'm posting links to two papers without comment. One of them is from my friend and colleague Alex Palazzo and the other is from James Shapiro who is not my friend or colleague. Both papers have been published in reputable peer-review journals.

A detailed examination of the new complete human genome reveals that 54% of it consists of various repetitive elements. Some of them are transcribed and some aren't.
This is my fourth post on the complete telomere-to-telomere sequence of the human genome in cell line CHM13 (T2T-CHM13). There were six papers in the April 1st edition of Science. My posts on all six papers are listed at the bottom of this post.
The fourth paper extends the ENCODE-type analysis of the T2T-CHM13 sequence by focusing on repeats.
Hoyt, S.J., Storer, J.M., Hartley, G.A., Grady, P.G., Gershman, A., de Lima, L.G., Limouse, C., Halabian, R., Wojenski, L., Rodriguez, M. et al. (2021) From telomere to telomere: the transcriptional and epigenetic state of human repeat elements. Science 376:57. [doi: 10.1126/science.abk3112]
Mobile elements and repetitive genomic regions are sources of lineage-specific genomic innovation and uniquely fingerprint individual genomes. Comprehensive analyses of such repeat elements, including those found in more complex regions of the genome, require a complete, linear genome assembly. We present a de novo repeat discovery and annotation of the T2T-CHM13 human reference genome. We identified previously unknown satellite arrays, expanded the catalog of variants and families for repeats and mobile elements, characterized classes of complex composite repeats, and located retroelement transduction events. We detected nascent transcription and delineated CpG methylation profiles to define the structure of transcriptionally active retroelements in humans, including those in centromeres. These data expand our insight into the diversity, distribution, and evolution of repetitive regions that have shaped the human genome.

The most useful part of this paper is the complete analysis of all repetitive elements in the T2T-CHM13 genome. This gives us, for the first time, a complete picture of a human genome. The exact values of the various components aren't important because there's considerable variation with the human population but the big picture is informative.
These are the percentages of the human genome occupied by the different classes of repetitive DNA.
- SINEs 12.8%
- Retrotransposon 0.15%
- LINEs 20.7%
- LTRs 8.8%
- DNA transposons 3.6%
- simple repeats 8%
The total comes to 54%. There are other estimates that are higher because of a more lenient cutoff value for sequence similarity but this gives you a pretty good idea of what the genome looks like. Most of the transposon-related sequence consists of fragments of once active transposons so the fraction of the genome consisting of true selfish DNA capable of transposing is a small fraction of this 54%.
We have every reason to believe that most of this DNA is junk DNA based on several lines of evidence developed over the past 50 years but most of the authors of this paper are reluctant to reach that conclusion so the fact that these repetitive sequences might be junk isn't mentioned in the paper. Instead, the authors concentrate on mapping CpG methylation sites and transcribed regions. They refer to this as "functional annotation" but they don't provide a definition of function.
We provide a high-confidence functional annotation of repeats across the human genome.
As you might expect, the repeat elements that retain vestiges of promoters are often transcribed and this includes adjacent genomic sequences that are found near these promoter (e.g. near LTRs). The long stretches of short tandem repeats (e.g. satellite DNA) do not contain any sequences that resemble promoters so these regions are not transcribed. (The authors seem to be a bit surprised by this result.) Further work is needed to decide how much of this DNA is truly functional and which parts contribute to human uniqueness. Naturally, that will require much more ENCODE-type work and T2T sequencing of other primates.
Finally, our work demonstrates the need to increase efforts toward achieving T2T-level assemblies for nonhuman primates to fully understand the complexity and impact of repeat-derived genomic innovations that define primate lineages, including humans. Although we find repeat variants that appear enriched or specific to the human lineage, in the absence of T2T-level assemblies from other primate species, we cannot truly attribute these elements to specific human phenotypes. Thus, the extent of variation described herein highlights the need to expand the effort to create human and nonhuman primate pan-genome references to support exploration of repeats that define the true extent of human variation.
This will cost millions of dollars. I suspect the grant applications have already been sent.

Check out this freshperson seminar course on Parts Unknown: The Dark Matter of the Genome at Harvard. It is offfered by Amanda J. Whipple of the Department of Molecular and Cellular Biology. She works on noncoding RNAs in the brain. Harvard likes to think of itself as one of the top universities in the world so this seminar course must be an example of world class critical thinking.
Heaven help us if this is what future American leaders are being taught.
Did you know that genes, traditionally defined as DNA encoding protein, only account for two percent of the entire human genome? What is the purpose of the remaining 98% of the genome? Is it simply “junk DNA”? This seminar will explore the large portion of our genome that has been neglected by scientists for many years because its purpose was not known. We will examine research findings which demonstrate non-coding sequences, previously assigned as “junk DNA”, play crucial roles in the development and maintenance of a healthy organism. We will further discuss how these non-coding sequences are promising targets for drug design and disease diagnosis. We will then visit a local research laboratory (either virtually or in person as deemed appropriate) and engage with active scientists regarding the scientific research enterprise.
A thorough understanding of the human genome not only provides a foundation for any student interested in the life sciences, it enables one to engage more deeply in related political and societal debates, which is expected to become even more central as scientists further uncover the dark matter of our genomes.
Setting aside the sarcasm, how did we get to a stage where a prominent researcher at one of the top research universities in the world could write such a course description?

Karen Miga deserves a lot of the credit for the complete human genome sequence.
Karen Miga is a professor at the University of California, Santa Cruz, and she's been working for several years on sequencing the repetitive regions of the genome. She is a co-founder of the telomere-to-telomere consortium that just published a complete sequece of the human genome. She made a signficant contribution to long-read (~20 Kb) and ultra-long-read (>100 kb) sequencing and that's a major technological achievement that's worthy of prizes.
Read the interview on CBC (Canada) Quirks & Quarks at Scientists sequence complete, gap-free human genome for the first time and watch the YouTube video.
Miga did her Ph.D. with Huntington Willard at Duke University. Hunt has been working on centromeres for more than 40 yeas years and some of my colleagues may remember him when he was a professor at the University of Toronto in the Department of Medical Genetics.

It's going to be extremely difficult, perhaps impossible, to merge the new complete human genome sequence with the current standard reference genome.
The source DNA for the new telomere-to-telomere (T2T) human genome sequence was a cell line derived from a molar pregnancy. This meant that the DNA was essentially haploid, thus avoiding the complications of sequencing diploid DNA which contains two highly similar but different genomes. The cell line, CHM13, lacks a Y chromosome but that's trivial since a complete T2T sequence of a Y chromosome will soon be published and it can be added to the T2T-CHM13 genome sequence [Telomere-to-telomere sequencing of a complete human genome].

The new completed human genome sequence contains some previously unknown large duplicatons (segmental duplications).
This is my third post on the complete telomere-to-telomere sequence of the human genome in cell line CHM13 (T2T-CHM13). There were six papers in the April 1st edition of Science. My posts on all six papers are listed at the bottom of this post.

The newly sequenced part of the human genome contains the same chromatin regions as the rest of the genome and they don't tell us very much about which regions are functional and which ones are junk.
This is my second post on the complete telomere-to-telomere sequence of the human genome in cell line CHM13 (T2T-CHM13). There were six papers in the April 1st edition of Science. My posts on all six papers are listed at the bottom of this post.
The first complete human genome sequence has finally been published.
This is my first post on the complete telomere-to-telomere sequence of the human genome in cell line CHM13 (T2T-CHM13). There were six papers in the April 1st edition of Science. My posts on all six papers are listed at the bottom of this post.
A few months ago, the press office of the University of California at San Diego issued a press release with a provocative title ...
Illuminating Dark Matter in Human DNA - Unprecedented Atlas of the "Book of Life"
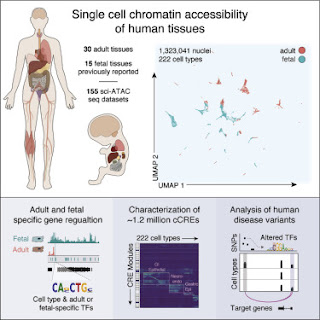
The press release was posted on several prominent science websites and Facebook groups. According to the press release, much of the human genome remains mysterious (dark matter) even 20 years after it was sequenced. According to the senior author of the paper, Bing Ren, we still don't understand how genes are expressed and how they might go awry in genetic diseases. He says,
A major reason is that the majority of the human DNA sequence, more than 98 percent, is non-protein-coding, and we do not yet have a genetic code book to unlock the information embedded in these sequences.
We've heard that story before and it's getting very boring. We know that 90% of our genome is junk, about 1% encodes proteins, and another 9% contains lots of functional DNA sequences, including regulatory elements. We've known about regulatory elements for more than 50 years so there's nothing mysterious about that component of noncoding DNA.

John Mattick and Paulo Amaral have written a book that promotes their views on the content of the human genome. It will be available next August. Their main thesis is that the human genome is full of genes for regulatory RNAs and there's very little junk. A secondary theme is that some very smart scientists have been totally wrong about molecular biology and molecular evolution for the past fifty years.
I pretty much know what's going to be in the book [see John Mattick presents his view of genomes]. I also know that most of his claims don't stand up to close scrutiny but that's not going to prevent it from being touted as a true paradigm shift. (It's actually a paradigm shaft.) I suspect it's going to get favorable reviews in Science and Nature.
John Mattick has a new book coming out in August where he defends the notion that most of our genome is full of genes for functonal noncoding RNAs. We have a pretty good idea what he's going to say. This is a talk he gave at Oxford on May 17, 2019.
Here are a few statements that should pique your interest.
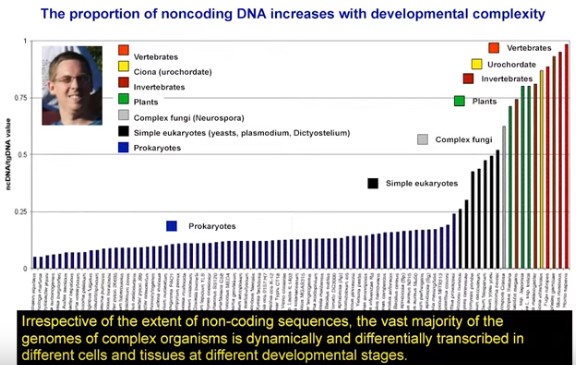

I don't know if there were questions but, if there were, I bet that none of them challenged Mattick's main thesis.

Science editors asked young scientists to imagine what kind of course they would have created if they could go back to a time before the pandemic [A pandemic education]. Three of the courses were about science communication.
COM 145: Identification, analysis, and communication of scientific evidence
This course focuses on developing the skills required to translate scientific evidence into accessible information for the general public, especially under circumstances that lead to the intensification of fear and misinformation. Discussions will cover the principles of the scientific method, as well as its theoretical and practical relevance in counteracting the dissemination of pseudoscience, particularly on social media. This course discusses chapters from Carl Sagan’s book The Demon-Haunted World, certain peer-reviewed and retracted papers, and materials related to key science issues, such as the anti-vaccine movement. For the final project, students will comprehensibly communicate a scientific topic to the public.
Camila Fonseca Amorim da Silva University of Sao Paulo, Sao Paulo, Brazil
COM 198: Everyday science communication
As scientific discoveries become increasingly specialized, the lack of understanding by the general public undermines trust in scientists and causes the spread of misinformation. This course will be taught by scientists and communication specialists who will provide students with a toolset to explain scientific concepts, as well as their own research projects, to the general public. Upon completion of this course, students will be able to explain to their grandparents that viruses exist even though they can’t see them, convince their neighbors that vaccines don’t contain tracking devices, and explain the concept of exponential growth to governmental officials.
Anna Uzonyi Department of Molecular Genetics, Weizmann Institute of Science, Rehovot, Israel.
COM 232: Introduction to talking to regular people
Communicating science is difficult. Many scientists, having immersed themselves in the language of their field, have completely forgotten how to talk to regular people. This course hones introductory science communication skills, such as how to talk about scary things without generating mass panic, how to calmly discourage the hoarding of paper hygiene products, and how to explain why scientific knowledge changes over time. The final project will include cross examination from law school faculty, who are otherwise completely uninvolved with the course and possess minimal scientific training. Recommended for science majors who are unable to discuss impactful scientific findings without citing a P value.
Joseph Michael Cusimano Bernard J. Dunn School of Pharmacy, Shenandoah University, Winchester, VA, USA.
They sound like interesting courses but my own take on science communication is somewhat different. I think it's very difficult for practicing scientists to communicate effectively with the general public so I tend to view science communication at several different levels. My goal is to communicate with an audience of scientists, science journalists, and people who are already familiar with science. The idea is to make sure that this intermediate group understands the scientific facts in my field and to make sure they are familiar with the major controversies.
My hope is that this intermediate group will disseminate this information to their less-informed friends and relatives and, more importantly, stop the spread of misinformation whenever they hear it.
Take junk DNA for example. It's very difficult to convince the average person that 90% of our genome is junk because the idea is so counter-intuitive and contrary to the popular counter-narratives. However, I have a chance of convincing the intermediate group, including science journalists and other scientists, who can follow the scientific arguments. If I succeed, they will at least stop spreading misinformation and false narratives and start presenting alternatives to their sudiences.

My book manuscript has been reviewed by some outside experts and they seem to have convinced my editor that my book is worth publishing. I hope we can get it finished soon. It would be nice to publish in in September on the 10th anniversary of the ENCODE disaster.
Meanwhile, I keep scanning the literature for mentions of junk DNA to see if scientists are finally coming to their senses. Apparently not, and that's a good thing because it means that my book is still needed. Here's the opening paragraph from a recent review of lncRNAs. The authors are in the Department of Medicine at the Medical College of Gerogia, in Augusta, Georgia (USA).
Ghanam, A.R., Bryant, W.B. and Miano, J.M. (2022) Of mice and human-specific long noncoding RNAs. Mammalian Genome:1-12. [doi: 10.1007/s00335-022-09943-2]
Approximately ninety-eight percent of our genome is noncoding. Contrary to initial descriptions of this vast sea of sequence comprising “junk DNA” (Ohno 1972), comparative genomics and various next-generation sequencing studies have revealed millions of transcription factor binding sites (TFBS) (Vierstra et al. 2020) and tens of thousands of noncoding genes, most notably the class of long noncoding RNAs (LncRNAs), defined currently as processed transcripts of length > 200 base pairs with no protein-coding capacity (Rinn and Chang 2020; Statello et al. 2021). The widespread transcription of LncRNAs and abundance of regulatory sequences such as enhancers support the concept of a genome that is largely functional (ENCODE Project Consortium 2012). Such a dynamic genome should not be surprising given the complex nature of gene expression and gene function necessary for embryonic and postnatal development as well as disease processes.
Except for those few minor details—I hope I'm not being too picky—that's a pretty good way to start a review of lncRNAs. :-)

Jacques Fresco died last December. I am kind of a scientific grandson of Jacques Fresco since he mentored my Ph.D. supervisor, Bruce Alberts when he (Bruce) was an undergraduate at Harvard.
While at Harvard, Jacques mentored then-undergraduate Bruce Alberts, who taught at Princeton from 1966 to 1976, served as president of the National Academy of Sciences and wrote the seminal textbook, “The Molecular Biology of the Cell.”In addition to reassuring Alberts’ parents that they shouldn’t worry about their son’s choice to pursue science instead of medical school — a story Fresco enjoyed telling — he also played a key role in bringing the young scientist to Princeton. “Before I had even completed my Ph.D., he convinced Princeton to offer me an assistant professorship that I did not deserve,” Alberts recalled. “And at Princeton for 10 years, we of course spent an enormous amount of time together. So Jacques was very central to my life as a scientist and a close friend.”
The Fresco lab was right above the Alberts lab when I started graduate school at Princeton in 1968. The main focus of the Fresco lab was the structure of tRNA and in order to isolate different tRNA molecules they needed a very large gel filtration column that was about 4m tall and about as big around as a dinner plate. The column was too tall for their lab so they had to drill a hole through the concrete floor and drop it down into the lab below!
One of my graduate student friends worked in the Fresco lab on hydrogen exchange in tRNA. The idea was to measure the number of hydrogen bonds in the structure by looking at the exchange bewtween hydrogens in the medium and in tRNA. The experiment used a radioactive isotope of hydrogen (tritium) in the medium and each experiment required about one curie of radioactive hydrogen and that's a lot. After a few years my friend decided to become a plastic surgeon instead of a scientist!
I knew Jacques Fresco quite well when I was a graduate student and I always thought he was an excellent scientist.Many of his students mentioned what an enormous role Fresco played in shaping their careers, in large and small ways. “Jacques treated everybody with the same respect, irreverence and love of life,” said Steven Broitman, a professor of biology at West Chester University in Pennsylvania who completed his Ph.D. with Fresco in 1988. “In addition to all he taught me about science, he also modeled the simple enjoyment in doing science that I have always tried to keep with me and pass on to my own students. He was larger than life, a major figure in the birth of modern molecular biology. He was deeply loved, and he will be missed.”