A few months ago, the press office of the University of California at San Diego issued a press release with a provocative title ...
Illuminating Dark Matter in Human DNA - Unprecedented Atlas of the "Book of Life"
The press release was posted on several prominent science websites and Facebook groups. According to the press release, much of the human genome remains mysterious (dark matter) even 20 years after it was sequenced. According to the senior author of the paper, Bing Ren, we still don't understand how genes are expressed and how they might go awry in genetic diseases. He says,
A major reason is that the majority of the human DNA sequence, more than 98 percent, is non-protein-coding, and we do not yet have a genetic code book to unlock the information embedded in these sequences.
We've heard that story before and it's getting very boring. We know that 90% of our genome is junk, about 1% encodes proteins, and another 9% contains lots of functional DNA sequences, including regulatory elements. We've known about regulatory elements for more than 50 years so there's nothing mysterious about that component of noncoding DNA.
Ren and his colleagues are trying to identify regulatory elements by looking at transcription factor binding sites and their associated chromatin alterations. If this sounds familiar, it's because scientists have been mapping these sites for at least 25 years.
Efforts to fill in the blanks are broadly captured in an ongoing international effort called the Encyclopedia of DNA Elements (ENCODE), and include the work of Ren and colleagues. In particular, they have investigated the role and function of chromatin, a complex of DNA and proteins that form chromosomes within the nuclei of eukaryotic cells.
Back in 2007 and 2012, ENCODE started mapping all the spurious transcription factor binding sites in the human genome. They also mapped all the spurious transcripts that are due to nonfunctional transcription and all the open chromatin domains associated with those sites. There are millions of these sites and most of them have nothing to do with normal gene expression and biological function.
The paper being promoted by the press release (Zhang et al., 2021) continues this work by mapping more spurious transcription factor binding sites. A tiny percentage of these might be genuine cis-regulatory elements (cCREs).
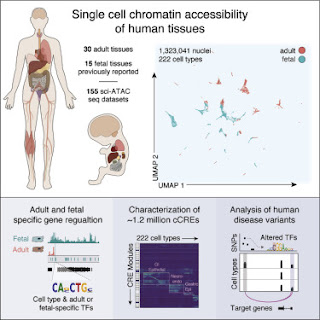
They applied assays to more than 600,000 human cells sampled from 30 adult human tissue types from multiple donors, then integrated that information with similar data from 15 fetal tissue types to reveal the status of chromatin at approximately 1.2 million candidate cis-regulatory elements in 222 distinct cell types.
The paper reports a total of 1,154,661 "distinct cCREs" spanning 14.8% of the genome. In typical fashion, there's no mention of spurious binding sites and very little attempt to identify real regulatory sites. As usual, all the sites are called cis-regulatory elements even though the authors must surely know that most of them are NOT regulatory elements. Most of that 14.8% of the genome is junk DNA but the authors forget to mention that possibility. I'm sure it just slipped their mind because they must know about spurious sites after all the controversy over earlier ENCODE results dating back to 2007.
Much of the emphasis in the paper is on the possible association of these sites with various diseases. Here's what they say in the introduction.
Genome-wide association studies (GWAS) have identified hundreds of thousands of genetic variants associated with a broad spectrum of human traits and diseases. The large majority of these variants are noncoding.
That's an interesting piece of information. If true, it means that there are at least 200,000 "genetic variants" or about eight per gene. Most of them will be in junk DNA and will have nothing to do with any nearby genes. They just happen to be linked to those genes. The authors identified 527 transcription factor binding sites that might possibly be associated with genetic variants that could be influencing nearby genes. Two of them look promising; one is associated with ulcerative colitis and one is associated with osteoarthritis. That's two out of 1.2 million.
I think this is what they mean when they say they're illuminating the dark matter of the genome.
Nobody questions the usefullness of GWAS in mapping genes to phenotypes, including diseases. Nobody doubts that some of these genetic association studies will actually reveal the genetic cause of the phenotypic change, as opposed to fortuitous associations with linked polymorphisms. Nobody is surprised that most of these genetic markers lie outside of protein-coding regions. What surprises me is that the benefits of these mapping experiments aren't presented in a context that recognizes the limitations and the difficulties in interpreting the data. In this case, why not discuss the fact that the vast majority of these so-called cCREs are likely to be spurious transcription factor binding sites that have nothing to do with gene regulation or disease? This means that the problem of identifying a few functional promoters amid a sea of irrelevant features is very difficult. Why not say that?
I struggle to come up with an explanation for this behavior. Is it because the authors know about spurious binding sites but don't want to complicate their paper by mentioning them? Or is it because the authors don't really understand junk DNA and spurious binding sites? And how do you explain the reviewers who approve these papers for publication?
Zhang, K., Hocker, J.D., Miller, M., Wang, A., Preisel, S. and Ren, B. (2021) A single-cell atlas of chromatin accessibility in the human genome. Cell 184: 5985-6001 e5919 doi: 10.1016/j.cell.2021.10.024
10 comments :
A naive question for the commentariat if you will. Is the 'junk' component ratio changing over time? What are possible conceptual reasons for 'junk' existence? It seems that its existence implies some drag on the organism. Or does junk simply mean not useful for this particular purpose?
The junk ratio does change over time. Some genomes get bigger and some get smaller, and almost all size change can be attributed to gain or loss of junk sequences. There is seldom any drag, if by that you mean negative selection. DNA replication just isn't a very large part of a cell's energy budget. And "junk" refers to DNA that does nothing, not useful for any current purpose.
Thanks for replying. I'll take it as the conventional/consensus view.
So then it appears that selection exists against junk but is not strong b/c energy demand for it is relatively low. How to explain junk's large %? Errors,chance?
It's not easy to define the "consensus view." If we are being honest, then I think it's fair to say that the consensus view is very different from the one I am advocating. Unfortunately, it seems like the vast majority of molecular biologists think that most of our genome is functional. You can see evidence of this in press releases, popular books and articles, and even in papers that are being published every day in the scientific literature.
The view that 90% of our genome is junk is being completely ignored. It's not even mentioned as a plausible hypothesis that's supposedly rejected by the "evidence."
So non-coding DNA is junk from your viewpoint. Because it is non coding or junk for any purpose? From this perspective is junk a drag?
I tend (prefer) to a view that bio systems are essentially deterministic and respond to energy inputs/distribution. If this is plausible how to account for the continued thriving junk? Competition? Randomness does not fit well with my bias.
a view that bio systems are essentially deterministic and respond to energy inputs/distribution.
Well, that one's definitely not the consensus view of molecular evolutionists. They know a lot of processes that put junk into the genome and aren't well-described as "essentially deterministic" in that way. The consensus of molecular evolutionists is that there is somewhere about 90% of the genome that is "junk". As Larry notes, the majority, maybe at least 2/3 of genomicists and molecular biologists think that most of the genome is "functional". There are more of them than there are of us, so I guess their view is "the consensus".
I am a lay person and I am not advancing a serious argument. I may have inadvertently overemphasized 'consensus' trying to avoid needless controversy.
If the view is not mostly deterministic then what?
My preference is immaterial to the discussion, I know, but it is there for reasons I consider rational. FWIW the medical technology (science?) in my view and experience is ineffective and naive in its broad assumption that life do not behave like a dynamic system rather a bunch of broken parts. Biology is guilty by association?
Personal experience suggests that my body behaves like a low entropy system, with some reversal possible (not unexpected). So my personal experience is surprisingly better than expected. I am not selling anything.
Morris, I suggest you learn about transposons. They are parasitic bits of DNA which can get themselves copied within a genome. They are somewhat analogous to infectious diseases, though they only transmit down the generations, and operate over very long (evolutionary) time scales. They can make a lot of junk. Do you think that infectious diseases are mostly deterministic?
I think there is so much noise that it is only possible to discern broad patterns. I think questions like "why do bats harbour more viruses than other mammals?" or "why do bats have less junk than other mammals?" can be answered, but it's very tricky when it's this species vs that species.
So then it appears that selection exists against junk but is not strong b/c energy demand for it is relatively low. How to explain junk's large %? Errors,chance?
Yes, errors, chance. Mutations fixed by drift. Now in fact there might be selection against junk, but too weak to have any effect. There is a rough negative correlation between population size and the amount of junk, which suggests some negative selection. One problem for selection is that it can't select against junk in general, only against differences in the amount of junk among individuals in the population. And this amount varies much more among species than within species. Could species selection influence the number of species with lots of junk? Maybe. But based on the distribution of genome sizes inthe biota, not much.
@GJ
Thanks for your suggestion. I have read Robert Triver's book on transposons. I enjoyed the book but did not understand it, just took it as interesting. To say I understand something I want to be able to think of alternate (conceptual) possibilities and to see gaps (what is not mentioned) in the proposal. Much too late for me in this field.
Acute infections seem to be deterministic if I correctly understand empirical measures of antigen/antibody variation. What is more interesting (and important personally) is the chronic state infections or disease of aging. Why so little , if any debate on this topic? Why is coevolution/competition of species in the human body ecology not widely discussed? I think these are gaps which ought to be examined. Id it b/c it's a dead end?
Surely more important personally than exact ratios of junk:coding.
Post a Comment