A hard-hitting review will be published in Annual Review of Genomics and Human Genetics. It shows that the case for large numbers of functional lncRNAs is grossly exaggerated.
A long-time Sandwalk reader (Ole Kristian Tørresen) alerted me to a paper that's coming out next October in Annual Review of Genomics and Human Genetics. (Thank-you Ole.) The authors of the review are Chris Ponting from the University of Edinburgh (Edinburgh, Scotland, UK) and Wilfried Haerty at the Earlham Institute in Norwich, UK. They have been arguing the case for junk DNA for the past two decades but most of their arguments are ignored. This paper won't be so easy to ignore because it makes the case forcibly and critically reviews all the false claims for function. I'm going to quote a few juicy parts because I know that many of you will not be able to access the preprint.
Among other things, Ryan picks on the views of John Mattick who has got to be one of the worst scientists in the field. Whenever I read a paper by Mattick I revise my opinion of the value of peer-reviewed literature. It's bad enough that Mattick has silly ideas but it's even sadder that his "peer" reviewers don't recognize it.
Here's a quote from Mattick that I discussed in my article on the The Central Dogma of Molecular Biology. It's obvious that he doesn't understand the real meaning of the central dogma. Can you pick out the other conceptual flaws in this paragraph? [Hints: Worst Figure Ever and Dog Ass Plots.]
The central dogma of biology holds that genetic information normally flows from DNA to RNA to protein. As a consequence it has been generally assumed that genes generally code for proteins, and that proteins fulfil not only most structural and catalytic but also most regulatory functions, in all cells, from microbes to mammals. However, the latter may not be the case in complex organisms. A number of startling observations about the extent of non-protein coding RNA (ncRNA) transcription in the higher eukaryotes and the range of genetic and epigenetic phenomena that are RNA-directed suggests that the traditional view of genetic regulatory systems in animals and plants may be incorrect.
Mattick, J.S. (2003) Challenging the dogma: the hidden layer of non-protein-coding RNAs in complex organisms. BioEssays 25:930-939.
Nature Methods is one of the journals in Nature Portfolio published by Springer Nature. Its focus is novel methods in the life sciences.
The latest issue (October, 2022) highlights the issues with identifying functional noncoding RNAs and the editorial, Decoding noncoding RNAs, is quite good—much better than the comments in other journals. Here's the final paragraph.
Despite the increasing prominence of ncRNA, we remind readers that the presence of a ncRNA molecule does not always imply functionality. It is also possible that these transcripts are non-functional or products from, for example, splicing errors. We hope this Focus issue will provide researchers with practical advice for deciphering ncRNA’s roles in biological processes.
However, this praise is mitigated by the appearance of another article in the same journal. Science journalist, Vivien Marx has written a commentary with a title that was bound to catch my eye: How noncoding RNAs began to leave the junkyard. Here's the opening paragraph.
Junk. In the view of some, that’s what noncoding RNAs (ncRNAs) are — genes that are transcribed but not translated into proteins. With one of his ncRNA papers, University of Queensland researcher Tim Mercer recalls that two reviewers said, “this is good” and the third said, “this is all junk; noncoding RNAs aren’t functional.” Debates over ncRNAs, in Mercer’s view, have generally moved from ‘it’s all junk’ to ‘which ones are functional?’ and ‘what are they doing?’
This is the classic setup for a paradigm shaft. What you do is create a false history of a field and then reveal how your ground-breaking work has shattered the long-standing paradigm. In this case, the false history is that the standard view among scientists was that ALL noncoding RNAs were junk. That's nonsense. It means that these old scientists must have dismissed ribosomal RNA and tRNA back in the 1960s. But even if you grant that those were exceptions, it means that they knew nothing about Sidney Altman's work on RNAse P (Nobel Prize, 1989), or 7SL RNA (Alu elements), or the RNA components of spliceosomes (snRNAs), or PiWiRNAs, or snoRNAs, or microRNAs, or a host of regulatory RNAs that have been known for decades.
Knowledgeable scientists knew full well that there are many functional noncoding RNAS and that includes some that are called lncRNAs. As the editorial says, these knowledgeable scientists are warning about attributing function to all transcripts without evidence. In other words, many of the transcripts found in human cells could be junk RNA in spite of the fact that there are also many functional nonciding RNAs.
So, Tim Mercer is correct, the debate is over which ncRNAs are functional and that's the same debate that's been going on for 50 years. Move along folks, nothing to see here.
The author isn't going to let this go. She decides to interview John Mattick, of all people, to get a "proper" perspective on the field. (Tim Mercer is a former student of Mattick's.) Unfortunately, that perspective contains no information on how many functional ncRNAs are present and on what percentage of the genome their genes occupy. It's gonna take several hundred thousand lncRNA genes to make a significant impact on the amount of junk DNA but nobody wants to say that. With John Mattick you get a twofer: a false history (paradigm strawman) plus no evidence that your discoveries are truly revolutionary.
Nature Methods should be ashamed, not for presenting the views of John Mattick—that's perfectly legitimate—but for not putting them in context and presenting the other side of the controversy. Surely at this point in time (2022) we should all know that Mattick's views are on the fringe and most transcripts really are junk RNA?
I'm discussing a recent paper published by Nils Walter (Walter, 2024). He is trying to explain the conflict between proponents of junk DNA and their opponents. His main focus is building a case for large numbers of non-coding genes.
This is the second post in the series. The first one outlines the issues that led to the current paper.
Walter begins his defense of function by outlining a "paradigm shift" that's illustrated in Figure 1.
FIGURE 1: Assessment of the information content of the human genome ∼20 years before (left)[110] and after (right)[111] the Human Genome Project was preliminarily completed, drawn roughly to scale.[9] This significant progress can be described per Thomas Kuhn as a “paradigm shift” flanked by extended periods of “normal science”, during which investigations are designed and results interpreted within the dominant conceptual frameworks of the sub-disciplines.[9] Others have characterized this leap in assigning newly discovered ncRNAs at least a rudimentary (elemental) biochemical activity and thus function as excessively optimistic, or Panglossian, since it partially extrapolates from the known to the unknown.[75] Adapted from Ref. [9].
Reference #9 is a paper by John Mattick promoting a "Kuhnian revolution" in molecular biology. I've already discussed that paper as an example of a paradigm shaft, which is defined as a strawman "paradigm" set up to make your work look like revolutionary [John Mattick's new paradigm shaft]. Here's the figure from the Mattick paper.
The Walter figure is another example of a paradigm shaft—not to be confused with a real paradigm shift.1 Both pie charts misrepresent the amount of functional DNA since they don't show regulatory sequences, centromeres, telomeres, origins of replication, and SARS. Together, these account for more functional DNA than the functional regions of protein-coding genes and non-coding genes. We didn't know the exact amounts in 1980 but we sure knew they existed. I cover this in Chapter 5 of my book: "The Big Picture."
The 1980 view also implies, incorrectly, that we knew nothing about the non-functional component of the genome when, in fact, we knew by then that half of our genome was composed of transposon and viral sequences that were likely to be inactive, degenerate fragments of once active elements. (John Mattick's figure is better.)
The 2020 view implies that most intron sequences are functional since introns make up more than 40% of our genome but only about 3% of the pie chart. As far as I know, there's no evidence to support that claim. About 80% of the pie chart is devoted to transcripts identified as either small ncRNAs or lncRNAs. The implication is that the discovery of these RNAs represents a paradigm shift in our understanding of the genome.
The alternative explanation is that we've known since the late 1960s that most of the human genome is transcribed and that these transcripts—most of which turned out to be introns—are junk RNA that is confined to the nucleus and rapidly degraded. Advances in technology have enabled us to detect many examples of spurious transcripts that are present transiently at low levels in certain cells. I cover this in Chaper 8 of my book: "Noncoding Genes and Junk RNA.
The whole point of Nils Walter's paper is to defend the idea that most of these transcripts are functional and the alternative explanation is wrong. He's trying to present a balanced view of the controversy so he's well aware of the fact that some of us interpret the red part of the pie chart as spurious transcripts (junk RNA). If he's wrong, and I am right, then there's no paradigm shift.
You don't get to shift the paradigm all on our own, even if John Mattick is on your side. A true paradigm shift requires that the entire community of scientists changes their perspective and that hasn't happened.
In the next few posts we'll see whether Nils Walter can make a strong case that all those lncRNAs are functional. They cover about two-thirds of the genome in the pie chart. If we assume that the average length of these long transcripts is 2000 bp then this represents one million transcripts and potentially one million non-coding genes.
1. The term "paradigm shaft" was coined by reader Diogenes in a comment on this blog from many years ago.
Walter, N.G. (2024) Are non‐protein coding RNAs junk or treasure? An attempt to explain and reconcile opposing viewpoints of whether the human genome is mostly transcribed into non‐functional or functional RNAs. BioEssays:2300201. [doi: 10.1002/bies.202300201]
Theme Genomes
& Junk DNAMany reputable scientists are convinced that most of our genome is junk. However, there are still a few holdouts and one of the most prominent is John Mattick. He believes that most of our genome is made up of thousand of genes for regulatory noncoding RNA. These RNAs (about 100 of them for every single protein-coding gene) are mostly involved in subtle controls of the levels of protein in human cells. (I'm not making this up. See: John Mattick on the Importance of Non-coding RNA )
It was a reasonable hypothesis at one point in time.
How do you evaluate a hypothesis in science? Well, one of the things you should always try to do is falsify your hypothesis. Let's see how that works ...
The RNAs should be conserved. FALSE
The RNAs should be abundant (>1 copy per cell). FALSE
There should be dozens of well-studied specific examples. FALSE
The hypothesis should account for variations in genome size. FALSE
The hypothesis should be consistent with other data, such as that on genetic load. FALSE
The hypothesis should be consistent with what we already know about the regulation of gene expression.FALSE
You should be able to refute existing hypotheses, such as transcription errors. FALSE
Normally, you would abandon a hypothesis that had such a bad track record but true believers aren't about to do that. So what's next? Maybe these regulatory RNAs don't show sequence conservation but maybe their secondary structures are conserved. In other words, these RNAs originated as functional RNAs with a secondary structure but over the course of time all traces of sequence conservation have been lost and only the "conserved" secondary structure remains.1 The Mattick lab looked at the "conservation" of secondary structure as an indicator of function using the latest algorithms (Smith et al., 2013). Here's how they describe their attempts to prove their hypothesis in light of conflicting data ...
The majority of the human genome is dynamically transcribed into RNA, most of which does not code for proteins (1–4). The once common presumption that most non–protein-coding sequences are nonfunctional for the organism is being adjusted to the increasing evidence that noncoding RNAs (ncRNAs) represent a previously unappreciated layer of gene expression essential for the epigenetic regulation of differentiation and development (5–8). Yet despite an exponential accumulation of transcriptomic data and the recent dissemination of genome-wide data from the ENCODE consortium (9), limited functional data have fuelled discourse on the amount of functionally pertinent genomic sequence in higher eukaryotes (1, 10–12). What is incontrovertible, however, is that evolutionary conservation of structural components over an adequate evolutionary distance is a direct property of purifying (negative) selection and, consequently, a sufficient indicator of biological function The majority of studies investigating the prevalence of purifying selection in mammalian genomes are predicated on measuring nucleotide substitution rates, which are then rated against a statistical threshold trained from a set of genomic loci arguably qualified as neutrally evolving (13, 14). Conversely, lack of conservation does not impute lack of function, as variation underlies natural selection. Given that the molecular function of ncRNA may at least be partially conveyed through secondary or tertiary structures, mining evolutionary data for evidence of such features promises to increase the resolution of functional genomic annotations.
Here's what they found ..
When applied to consistency-based multiple genome alignments of 35 mammals, our approach confidently identifies >4 million evolutionarily constrained RNA structures using a conservative sensitivity threshold that entails historically low false discovery rates for such analyses (5–22%). These predictions comprise 13.6% of the human genome, 88% of which fall outside any known sequence-constrained element, suggesting that a large proportion of the mammalian genome is functional.
Apparently 13.6% of the human genome is a "large proportion." Taken at face value, however, the Mattick lab has now shown that the vast majority of transcribed sequences don't show any of the characteristics of functional RNA, including conservation of secondary structure. Of course, that's not the conclusion they emphasize in their paper. Why not?
1. I can't imagine how this would happen, can you? You'd almost have to have selection AGAINST sequence conservation. Smith, M.A., Gese, T., Stadler, P.F. and Mattick, J.S. (2013) Widespread purifying selection on RNA structure in mammals. Nucleic Acid Research advance access July 11, 2013 [doi: 10.1093/nar/gkt596]
John Mattick continues to promote the idea that he is leading a paradigm shift in molecular biology. He believes that he and his colleagues have discovered a vast world of noncoding genes responsible for intricate gene regulation in complex eukaryotes. The latest salvo was fired a few months ago in June 2023.
Mattick, J.S. (2023) A Kuhnian revolution in molecular biology: Most genes in complex organisms express regulatory RNAs. BioEssays:2300080. [doi: 10.1002/bies.202300080]
Thomas Kuhn described the progress of science as comprising occasional paradigm shifts separated by interludes of ‘normal science’. The paradigm that has held sway since the inception of molecular biology is that genes (mainly) encode proteins. In parallel, theoreticians posited that mutation is random, inferred that most of the genome in complex organisms is non-functional, and asserted that somatic information is not communicated to the germline. However, many anomalies appeared, particularly in plants and animals: the strange genetic phenomena of paramutation and transvection; introns; repetitive sequences; a complex epigenome; lack of scaling of (protein-coding) genes and increase in ‘noncoding’ sequences with developmental complexity; genetic loci termed ‘enhancers’ that control spatiotemporal gene expression patterns during development; and a plethora of ‘intergenic’, overlapping, antisense and intronic transcripts. These observations suggest that the original conception of genetic information was deficient and that most genes in complex organisms specify regulatory RNAs, some of which convey intergenerational information.
This paper is promoted by a video in which he explains why there's a Kuhnian revolution under way. This paper differs from most of his others on the same topic because Mattick now seems to have acquired some more knowledge of the mutation load argument and the neutral theory of evolution. Now he's not only attacking the so-called "protein centric" paradigm but also the Modern Synthesis. Apparently, a slew of "anomalies" are casting doubt on several old paradigms.
This is still a paradigm shaft but it's a bit more complicated than his previous versions (see: John Mattick's paradigm shaft). Now his "anomalies" include not only large numbers of noncoding genes but also the C-value paradox, repetitive DNA, introns, enhancers, gene silencing, the g-value enigma, pervasive transcription, transvection, and epigenetics. Also, he now seems to be aware of many of the arguments for junk DNA but not so aware that he can reference any of his critics.1 His challenges to the Modern Synthesis include paramutation which, along with epigenetics, violate the paradigm of the Moden Synthesis because of non-genetic inheritance.
But the heart of his revolution is still the discovery of massive numbers of noncoding genes that only he and a few of his diehard colleague can see.
The genomic programming of developmentally complex organisms was misunderstood for much of the last century. The mammalian genome harbors only ∼20 000 protein-coding genes, similar in number and with largely orthologous functions as those in other animals, including simple nematodes. On the other hand, the extent of non-protein-coding DNA increases with increasing developmental and cognitive complexity, reaching 98.5% in humans. Moreover, high throughput analyses have shown that the majority of the mammalian genome is differentially and dynamically transcribed during development to produce tens if not hundreds of thousands of short and long non-protein-coding RNAs that show highly specific expression patterns and subcellular locations.
The figure is supposed to show that by 2020 junk DNA had been eliminated and almost all of the mammalian genome is devoted to functional DNA—mostly in the form of noncoding genes. There's only one very tiny problem with this picture—it's not supported by any evidence that all those functional noncoding genes exist. This is still a paradigm shaft of the third kind (false paradigm, false overthrow, false data).
1. There are 124 references; Dawkins and ENCODE make the list along with 14 of his own papers. Most of the papers in my list of Required reading for the junk DNA debate are missing. The absence of Palazzo and Gregory (2023) is particularly noteworthy.
John Mattick has a new book coming out in August where he defends the notion that most of our genome is full of genes for functonal noncoding RNAs. We have a pretty good idea what he's going to say. This is a talk he gave at Oxford on May 17, 2019.
Here are a few statements that should pique your interest.
(0:57) He says that his upcoming book is tentatively titled "the misunderstandings of molecular biology."
(1:11) He says that "the assumption has been very deeply embedded from the time of the lac operon on that genes equated to proteins."
(2:30) There have been three "surprises" in molecuular biology: (1) introns, (2) eukaryotic genomes are full of 'selfish' DNA, and (3) "gene number does not scale with developmental complexity."
(4:30) It is an unjustified assumption to assume that transposon-related seqences are junk and that leads to misinterpretation of neutral evolution.
(6:00) The view that evolution of regulatory sequences is mostly responsible for developmental complexity (Evo-Devo) has never been justified.
(8:45) A lot of obtuse theoretical discussion about how the number of regulatory protein-coding genes increases quadratically as the total number of protein-coding genes increase in a bacterial genome but at some point there has to be more protein-coding regulatory genes than total protein-coding genes so that limits the evolution of bacteria.
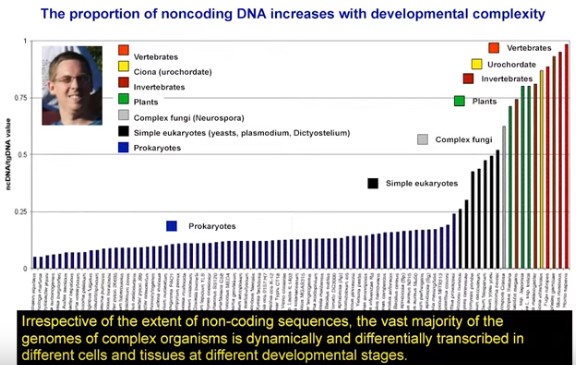
(13:40) The proportion of noncoding DNA increases with developmental complexity, topping out at humans.
(14:00) The vast majority of the genome in complex organisms is differentially transcribed in different cells and different tissues.
(14:15) The whole genome is alive on both strands.
(14:20) There are two possibilities: junk RNA or abundant functional transcripts and that explains complex organisms.
Mattick then takes several minutes to document the fact that there are abundant transcripts— a fact that has been known for the better part of sixty years but he does not mention that. All of his statements carry the implicit assumption that these transcripts are functional.
(20:20) He makes the boring, and largely irelevant, point that most disease-associated loci are located in noncoding regions (GWAS). He's responding to a critic who asked why, if these things (transcripts) are real, don't we see genetic evidence of it.
(24:00) Noncoding RNAs have all of the characteristics of functional RNAs with an emphasis on the fact that their expression is often only detected in specific cell types.
(31:50) It has now been shown that everything that protein transcription factors can do can be done by noncoding RNA.
(32:15) "I want to say to you that conservation is totally misunderstood." Apparently, lack of conservation imputes nothing about function.
(41:00) RNAs control phase separation. There's a whole other level of cell organization that we never dreamed of. (Ironically, he gives nucleoli as an example of something we never dreamed of.)
(42:36) "This is called soft metaphysics, and it's just come into biology, and it's spectacular in its implications."
(46:25) Almost every lncRNA is alternatively spliced in mice and humans.
(46:30) There's more alternative splicing in human protein-coding genes than in mice protein-coding genes but the extra splicing in humans is mostly in the 5' untranslated region. (I'm sure it has nothing to do with the fact that tons more RNA-Seq experiments have been done on human tissues.) "We think this is due to the increased sophistication of the regulation of these genes for the evolution of cognition."
(48:00) At least 20% of the human genome is evolutionarily conserved at the level of RNA structure and this does not require any assumptions.
(55:00) The talk ends at 55 minutes. That's too bad because I'm sure Mattick had a dozen more slides explaining why all of those transcripts are functional, as opposed to the few selected examples he picked. I'm sure he also had a lot of data refuting all of the evidence in favor of junk DNA but he just ran out of time.
I don't know if there were questions but, if there were, I bet that none of them challenged Mattick's main thesis.
All available evidence suggests that about 90% of our genome is junk DNA. Many scientists are reluctant to accept this evidence—some of them are even unaware of the evidence [Five Things You Should Know if You Want to Participate in the Junk DNA Debate]. Many opponents of junk DNA suffer from what I call The Deflated Ego Problem. They are reluctant to concede that humans have about the same number of genes as all other mammals and only a few more than insects.
One of the common rationalizations is to speculate that while humans may have "only" 25,000 genes they are regulated and controlled in a much more sophisticated manner than the genes in other species. It's this extra level of control that makes humans special. Such speculations have been around for almost fifty years but they have gained in popularity since publication of the human genome sequence.
In some cases, the extra level of regulation is thought to be due to abundant regulatory RNAs. This means there must be tens of thousand of extra genes expressing these regulatory RNAs. John Mattick is the most vocal proponent of this idea and he won an award from the Human Genome Organization for "proving" that his speculation is correct! [John Mattick Wins Chen Award for Distinguished Academic Achievement in Human Genetic and Genomic Research]. Knowledgeable scientists know that Mattick is probably wrong. They believe that most of those transcripts are junk RNAs produced by accidental transcription at very low levels from non-conserved sequences.
There's one post for each of the five issues that informed scientists need to address if they are going to write about the amount of junk in you genome. This is the last one.
There are several places in the book where Parrington address the issue of sequence conservation. The most detailed discussion is on pages 92-95 where he discusses the criticisms leveled by Dan Graur against ENCODE workers. Parrington notes that 9% of the human genome is conserved and recognizes that this is a strong argument for function. It implies that >90% of our genome is junk.
Here's how Parrington dismisses this argument ...
John Mattick and Marcel Dinger ... wrote an article for the HUGO Jounral, official journal of the Human Genome Organisation, entitled "The extent of functionality in the human genome." ... In response to the accusation that the apparent lack of sequence conservation of 90 per cent of the genome means that it has no function, Mattick and Dinger argued that regulatory elements and noncoding RNAs are much more relaxed in their link between structure and function, and therefore much harder to detect by standard measures of function. This could mean that 'conservation is relative', depending on the type of genomic structure being analyzed.
In other words, a large part of our genome (~70%?) could be producing functional regulatory RNAs whose sequence is irrelevant to their biological function. Parrington then writes a full page on Mattick's idea that the genome is full of genes for regulatory RNAs.
The idea that 90% of our genome is not conserved deserves far more serious treatment. In the next chapter (Chapter 7), Parrington discusses the role of RNA in forming a "scaffold" to organize DNA in three dimensions. He notes that ...
That such RNAs, by virtue of their sequence but also their 3D shape, can bind DNA, RNA, and proteins, makes them ideal candidates for such a role.
But if the genes for these RNAs make up a significant part of the genome then that means that some of their sequences are important for function. That has genetic load implications and also implications about conservation.
If it's not a "significant" fraction of the genome then Parrington should make that clear to his readers. He knows that 90% of our genome is not conserved, even between individuals (page 142), and he should know that this is consistent with genetic load arguments. However, almost all of his main arguments against junk DNA require that the extra DNA have a sequence-specific function. Those facts are not compatible. Here's how he justifies his position ...
Those proposing a higher figure [for functional DNA] believe that conservation is an imperfect measure of function for a number of reasons. One is that since many non-coding RNAs act as 3D structures, and because regulatory DNA elements are quite flexible in their sequence constraints, their easy detection by sequence conservation methods will be much more difficult than for protein-coding regions. Using such criteria, John Mattick and colleagues have come up with much higher figures for the amount of functionality in the genome. In addition, many epigenetic mechanisms that may be central for genome function will not be detectable through a DNA sequence comparison since they are mediated by chemical modifications of the DNA and its associated proteins that do not involve changes in DNA sequence. Finally, if genomes operate as 3D entities, then this may not be easily detectable in terms of sequence conservation.
This book would have been much better if Parrington had put some numbers behind his speculations. How much of the genome is responsible for making functional non-coding RNAs and how much of that should be conserved in one way of another? How much of the genome is devoted to regulatory sequences and what kind of sequence conservation is required for functionality? How much of the genome is required for "epigenetic mechanisms" and how do they work if the DNA sequence is irrelevant?
You can't argue this way. More than 90% of our genomes is not conserved—not even between individuals. If a good bit of that DNA is, nevertheless, functional, then those functions must not have anything to do with the sequence of the genome at those specific sites. Thus, regions that specify non-coding RNAs, for example, must perform their function even though all the base pairs can be mutated. Same for regulatory sequences—the actual sequence of these regulatory sequences isn't conserved according to John Parrington. This requires a bit more explanation since it flies on the face of what we know about function and regulation.
Finally, if you are going to use bulk DNA arguments to get around the conflict then tell us how much of the genome you are attributing to formation of "3D entities." Is it 90%? 70%? 50%?
One of the most important problems in biochemistry & molecular biology is the role (if any) of pervasive transcription. We've known for decades that most of the genome is transcribed at some time or other. In the case of organisms with large genomes, this means that tens of thousand of RNA molecules are produced from regions of the genome that are not (yet?) recognized as functional genes.
If you have a genome containing large amounts of junk DNA then it follows, as night follows day, that there will be a great deal of spurious transcription. The RNAs produced by these accidental events will not have a biological function.
I'm working on a chapter about pervasive transcription and how it relates to the junk DNA debate. I found a short review in Nature from 2002 so I decided to see how much progress we've made in the past 15 years.
Most of our genome is transcribed at some time or another in some tissue. That's a fact we've known about since the late 1960s (King and Jukes, 1969). We didn't know it back then, but it turns out that a lot of that transcription is introns. In fact, the observation of abundant transcription led to the discovery of introns. We have about 20,000 protein-coding genes and the average gene is 37.2 kb in length. Thus, the total amount of the genome devoted to these genes is about 23%. That's the amount that's transcribed to produce primary transcripts and mRNA. There are about 5000 noncoding genes that contribute another 2% so genes occupy about 25% of our genome.
Forty years ago it was thought that the amount of DNA in a genome correlated with the complexity of an organism. Back then, you often saw graphs like the one on the left. The idea was that the more complex the species the more genes it needed. Preliminary data seemed to confirm this idea.
In the late 1960's scientists started looking at the complexity of the genome itself. They soon discovered that large genomes were often composed of huge amounts of repetitive sequences. The amount of "unique sequence" DNA was only a few percent of the total DNA in these large genomes.1 This gave rise to the concept of junk DNA and the recognition that genome size was not a reliable indicator of the number of genes. That, plus the growing collection of genome size data, soon called into question the simplistic diagrams like the one shown here from an article by John Mattick in Scientific American (Mattick, 2004). (There are many things wrong with the diagram. Can you identify all of them? See What's wrong with this figure? at Genomicron).
Today we know that there isn't a direct correlation between genome size and complexity. Recent data, such as that from Ryan Gregory's website (right) reveals that the range of DNA sizes in many groups can vary over several orders of magnitude [Animal Genome Size Database]. Mammals don't have any more DNA in their genome than most flowering plants (angiosperms). Or even gymnosperms, for that matter.
Many of us have been teaching this basic fact for twenty years. The bottom line is ....
Anyone who states or implies that there is a significant correlation between total haploid genome size and species complexity is either ignorant or lying.
It is notoriously difficult to define complexity. That's only one of the reasons why such claims are wrong. Ryan Gregory wants everyone to know that the figure showing genome sizes in different phylogenetic groups is not meant to imply a hierarchy of complexity from algae to mammals.
A recent paper by Taft et al. (2007) says complexity can be "broadly defined as the number and different types of cells, and the degree of cellular organization." We can quibble about the definition but there's nothing better that I know of. The real question is whether organism complexity is a useful scientific concept.
Here's the problem. Have some scientists already made up their minds that mammals in general, and humans in particular, are the most complex organisms? Do they construct a definition f complexity that's guaranteed to confer the title of "most complex" on humans? Or, is complexity a real scientific phenomenon that hasn't yet been defined satisfactorily?
I, for one, don't know whether humans are more complex than an owl, or an octopus, or an orchid. For all I know, humans may be less complex by many scientific measure of complexity. Plants can grow and thrive on nothing but water, some minerals, and sunlight. We humans can't even make all of our own amino acids. Does that make us less complex than plants? Certainly it does at the molecular level.
Back in the olden days, when everyone was sure that humans were at the top of the complexity tree, the lack of correlation between genome size and complexity was called the C-value paradox where "C" stands for the haploid genome size. The term was popularized by Benjamin Lewin in his molecular biology textbooks. In Genes II (1983) he wrote.
The C value paradox takes its name from our inability to account for the content of the genome in terms of known function. One puzzling feature is the existence of huge variations in C values between species whose apparent complexity does not vary correspondingly. An extraordinary range of C values is found in amphibians where the smallest genomes are just below 109bp while the largest are almost 1011. It is hard to believe that this could reflect a 100-fold variation in the number of genes needed to specify different amphibians.
So, the paradox arises even if we don't know how to rank flowering plants and mammals of a complexity scale. It arises because there are so many examples of very similar species that have huge differences in the size of their genome. Onions, are another example—they are the reason why Ryan Gregory made up the Onion Test.
The onion test is a simple reality check for anyone who thinks they have come up with a universal function for non-coding DNA. Whatever your proposed function, ask yourself this question: Can I explain why an onion needs about five times more non-coding DNA for this function than a human?
Imagine the following scenario. You are absolutely convinced that humans are the most complex species but total genome size doesn't reflect your conviction. The C-value paradox is a real paradox for you. Knowing that much of our genome is possibly junk DNA still leaves room for plenty of genes. You take comfort in the fact that under all that junky genome, humans still have way more genes than simple nematodes and flowering plants. You were one of those people who wanted there to be 100,000 genes in the human genome [Facts and Myths Concerning the Historical Estimates of the Number of Genes in the Human Genome].
But when the genomes of these species are published, it turns out that even this faint hope evaporates. Humans, Arabidopsis (wall cress, right), and nematodes all have about the same number of genes.
Oops. Now we have a G-value paradox, where "G" is the number of genes (Hahn and Wray, 2002). The only way out of this box—without abandoning your assumption about humans being the most complex animals—is to make up some stories about the function of so-called junk DNA. If it turns out that there are lots of hidden genes in that junk then maybe it will rescue your assumption. This is where we get some combination of the excuses listed in The Deflated Ego Problem.
On the other hand, maybe humans really aren't all that much more complex, in terms of number of genes, than wall cress. Maybe they should have the same number of genes. Maybe the other differences in genome size really are due to variable amounts of non-functional junk DNA.
1. Thirty years ago we had to teach undergraduates about DNA reassociation kinetics and Cot curves—the most difficult thing I've ever had to teach. I'm sure glad we don't have to do that today.
Hahn, M.W. and Wray, G.A. (2002) The g-value paradox. Evol. Dev. 4:73-75.
Mattick, J.S. (2004) The hidden genetic program of complex organisms. Sci Am. 291:60-67.
Taft, R.J., Pheasant, M. and Mattick, J.S. (2007) The relationship between non-protein-coding DNA and eukarotic complexity. BioEssays 29:288-200.
[Photo Credits: The first figure is taken from a course webite at the University of Miami (Molecular Genetics. The second figure is from Ryan Gregory's Animal Genome Size Database (Statistics).]
"Pervasive transcription" refers to the idea that a large percentage of the DNA in mammalian genomes is transcribed. The idea became popular with the publication of the ENCODE results back in 2007 (Birney et al. 2007). Their results indicated that at least 93% of the human genome was transcribed at one time or another or in one tissue or another.
The result suggests that most of the genome consists of functional DNA. This pleases those who are opposed to the concept of junk DNA and it delights those who think that non-coding RNAs are going to radically change our concept of biochemistry and molecular biology. The result also pleased the creationists who were quick to point out that junk DNA is a myth [Junk & Jonathan: Part 6—Chapter 3, Most DNA Is Transcribed into RNA].
THEME: Transcription The original ENCODE paper used several different technologies to arrive at their conclusion. Different experimental protocols gave different results and there wasn't always complete overlap when it came to identifying transcribed regions of the genome. Nevertheless, the combination of results from three technologies gave the maximum value for the amount of DNA that was transcribed (93%). That's pervasive transcription.
The implication was that most of our genome is functional because it is transcribed.1 The conclusion was immediately challenged on theoretical grounds. According to our understanding of transcription, it is expected that RNA polymerase will bind accidentally at thousands of sites in the gnome and the probability of initiating the occasional transcript is significant [How RNA Polymerase Binds to DNA]. Genes make up about 30% of our genome and we expect that this fraction will be frequently transcribed. The remainder is transcribed at a very low rate that's easily detectable using modern technology. That could easily be junk RNA [How to Frame a Null Hypothesis] [How to Evaluate Genome Level Transcription Papers].
There were also challenges on technical grounds; notably a widely-discussed paper by van Bakel et al, 2010) from the labs of Ben Blencowe and Tim Hughes here in Toronto. That paper claimed that some of the experiments performed by the ENCODE group were prone to false positives [see Junk RNA or Imaginary RNA?]. They concluded,
We conclude that, while there are bona fide new intergenic transcripts, their number and abundance is generally low in comparison to known genes, and the genome is not as pervasively transcribed as previously reported.
The technical details of this dispute are beyond the level of this blog and, quite frankly, beyond me as well since I don't have any direct experience with these technologies. But let's not forget that aside from the dispute over the validity of the results, there is also a dispute over the interpretation.
As you might imagine, the pro-RNA, anti-junk, proponents fought back hard led by their chief, John Mattick, and Mark Gerstein (Clark et al., 2011). The focus of the counter-attack is on the validity of the results published by the Toronto group. Here's what Clark et al. (2011) conclude after their re-evaluation of the ENCODE results.
A close examination of the issues and conclusions raised by van Bakel et al. reveals the need for several corrections. First, their results are atypical and generate PR curves that are not observed with other reported tiling array data sets. Second, characterization of the transcriptomes of specific cell/tissue types using limited sampling approaches results in a limited and skewed view of the complexity of the transcriptome. Third, any estimate of the pervasiveness of transcription requires inclusion of all data sources, and less than exhaustive analyses can only provide lower bounds for transcriptional complexity. Although van Bakel et al. did not venture an estimate of the proportion of the genome expressed as primary transcripts, we agree with them that “given sufficient sequencing depth the whole genome may appear as transcripts” [2].
There is already a wide and rapidly expanding body of literature demonstrating intricate and dynamic transcript expression patterns, evolutionary conservation of promoters, transcript sequences and splice sites, and functional roles of “dark matter” transcripts [39]. In any case, the fact that their expression can be detected by independent techniques demonstrates their existence and the reality of the pervasive transcription of the genome.
The same issue of PLoS Biology contained a response from the Toronto group (van Bakel et al. 2011). They do not dispute the fact that much of the genome is transcribed since genes (exons + introns) make up a substantial portion and since cryptic (accidental) transcription is well-known. Instead, the Toronto group focuses on the abundance of transcripts from extra-genic regions and its significance.
We acknowledge that the phrase quoted by Clark et al. in our Author Summary should have read “stably transcribed”, or some equivalent, rather than simply “transcribed”. But this does not change the fact that we strongly disagree with the fundamental argument put forward by Clark et al., which is that the genomic area corresponding to transcripts is more important than their relative abundance. This viewpoint makes little sense to us. Given the various sources of extraneous sequence reads, both biological and laboratory-derived (see below), it is expected that with sufficient sequencing depth the entire genome would eventually be encompassed by reads. Our statement that “the genome is not as not as pervasively transcribed as previously reported” stems from the fact that our observations relate to the relative quantity of material detected.
Of course, some rare transcripts (and/or rare transcription) are functional, and low-level transcription may also provide a pool of material for evolutionary tinkering. But given that known mechanisms—in particular, imperfections in termination (see below)—can explain the presence of low-level random (and many non-random) transcripts, we believe the burden of proof is to show that such transcripts are indeed functional, rather than to disprove their putative functionality.
I'm with my colleagues on this one. It's not important that some part of the genome may be transcribed once every day or so. That's pretty much what you might expect from a sloppy mechanism—and let's be very clear about this, gene expression is sloppy.
You can't make grandiose claims about functionality based on such low levels of transcription. (Assuming the data turns out to be correct and there really is pervasive low-level transcription of the entire genome.)
This is a genuine scientific dispute waged on two levels: (1) are the experimental results correct? and (2) is the interpretation correct? I'm delighted to see these challenges to "dark matter" hyperbole and the ridiculous notion that most of our genome is functional. For the better part of a decade, Mattick and his ilk had free rein in the scientific literature [How Much Junk in the Human Genome?] [Greg Laden Gets Suckered by John Mattick].
We need to focus on re-educating the current generation of scientists so they will understand basic principles and concepts of biochemistry. The mere presence of an occasional transcript is not evidence of functionality and the papers that made that claim should never have gotten past reviewers.
1. Not just an "implication" since in many papers that conclusion is explicitly stated.
Birney, E., Stamatoyannopoulos, J.A. et al. (2007) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447:799-816. [doi:10.1038/nature05874]
van Bakel, H., Nislow, C., Blencowe, B. and Hughes, T. (2010) Most "Dark Matter" Transcripts Are Associated With Known Genes. PLoS Biology 8: e1000371 [doi:10.1371/journal.pbio.1000371]
van Bakel, H., Nislow, C., Blencowe, B.J., and Hughes, T.R.. (2011) Response to "the reality of pervasive transcription". PLoS Biol 9(7): e1001102. [doi:10.1371/journal.pbio.1001102]
The "Junk DNA" story is largely a myth, as you probably already know. DNA does not have to code for one of the few tens of thousands of proteins or enzymes known for any given animal, for example, to have a function. We know that. But we actually don't know a lot more than that, or more exactly, there is not a widely accepted dogma for the role of "non-coding DNA." It does really seem that scientists assumed for too long that there was no function in the DNA.
I hate to break it to you Greg, but junk DNA is not a myth. It really is true that a huge amount of our genome is junk. It's mostly defective transposons like SINES and LINES [Junk in your Genome: LINEs]. It's a lie that we don't know what most non-coding DNA is doing. We do know. It's not doing anything because it's mostly screwed up transposons and pseudogenes like Alu's.
Mattick may have found a few bits of DNA that encode regulatory RNAs but that's only a small part of the total genome. He, and you, have fallen for excuse #5 of The Deflated Ego Problem.
Human Evolution: Genes, Genealogies and Phylogenies was published in 2013 by Cambridge University Press. The author is Graeme Finlay, a cancer researcher at the University of Auckland, Auckland, New Zealand.
I first learned about this book from a book review published in the journal Evolution (Johnson, 2014). It sounded interesting so I bought a copy and read it.
There are four main chapters and each one covers a specific topic related to genomes and function. The topics are: Retroviruses, Transposons, Pseudogenes, and New Genes. There's lots and lots of interesting information in these chapters including an up-to-date summary of co-opted DNA that probably serves a biologically relevant function in our genome. This is the book to buy if you want a good review of the scientific literature on those topics.
Some of us have been trying to educate the IDiots for over twenty years. It can be very, very, frustrating.
The issue of junk DNA is a case in point. We've been trying to explain the facts to people like Casey Luskin. I know he's listening because he comments on Sandwalk from time to time. Surely it can't be that hard? All they have to do is acknowledge that "Darwinians" are opposed to junk DNA because they think that natural selection is very powerful and would have selected against junk DNA. All we're asking is that they refer to "evolutionary biologists" when they talk about junk DNA proponents.
We've also pointed out, ad nauseam, that no knowledgeable scientist ever said that all noncoding DNA was junk. We just want the IDiots to admit that there were some smart scientists who knew about functional noncoding DNA—like the genes for ribosomal RNAs, origins of replication, and centromeres.
I asked ChatGPT some questions about junk DNA and it made up a Francis Crick quotation and misrepresented the view of Susumu Ohno.
We have finally restored the Junk DNA article on Wikipedia. (It was deleted about ten years ago when Wikipedians decided that junk DNA doesn't exist.) One of the issues on Wikipedia is how to deal with misconceptions and misunderstandings while staying within the boundaries of Wikipedia culture. Wikipedians have an aversion to anything that looks like editorializing so you can't just say something like, "Nobody ever said that all non-coding DNA was junk." Instead, you have to find a credible reference to someone else who said that.
I've been trying to figure out how far the misunderstandings of junk DNA have spread so I asked ChatGPt (from OpenAI) again.
The latest issue of Nature contains a news feature by Anna Petherick [Genetics: The production line]. The article is mostly about a new regulatory RNA called HOTAIR but it's the implications of this discovery that bother me.
Let's look at the question being posed ...
If more than 90% of the genome is 'junk' then why do cells make so much RNA from it?
One of the answers being promoted by many molecular biologists is that this RNA is mostly functional and it represents a massive new level of control that has hitherto gone unnoticed. That doesn't mean that we never knew about regulatory RNAs because, in fact, we've known about them for over three decades. The important point of this opinion is that these RNAs are abundant and it "explains" the presence of so much non-coding DNA in eukaryotic genomes.
The other answer to the question is that transcription is sloppy and it frequently makes mistakes. That's why there's a very low level of transcription from all parts of the genome. It's junk RNA. This explanation seems to be widespread in the molecular biology community but it doesn't get much press because there are few papers that discuss this hypothesis [What is a gene, post-ENCODE?] [Junk RNA].
The probem in this field is that it's difficult to publish a paper that proves a negative (but see Brosius (2005)) and it's easy to publish a paper showing that a particular non-coding RNA has a function. The rare examples of those with function get all the publicity and obscure the fact that 99% of these transcripts may not have a function.
The article continues with ...
It is hard to comprehend the upheaval that RNA has been causing in molecular biology over the past few years. Once viewed as a passive intermediary, it was thought to faithfully carry genetic messages from the DNA sequence to the protein-making machinery, where things were made that actually got things done. Biologists were comfortable in the knowledge that only 1–2% of the human genome made protein-coding RNA in this way, and most of the rest was filler. So when, in 2005, geneticist Thomas Gingeras announced that some cells churn out RNA molecules from about 80% of their DNA, he astonished scientists attending the Biology of Genomes meeting at Cold Spring Harbor Laboratory in New York. Why should cells bother with so much manufacturing if, as it seemed, such a tiny fraction was involved in the important business of protein-making?
I wasn't at this meeting but I'd be very surprised if the scientists were "astonished." I'm pretty sure most of them thought that this was an artifact of some kind, probably due to accidental transcription.
This is a case where the author of the article could have benefited from interviewing more of the skeptics.
Over the past three years or so the case for this 'pervasive transcription' has strengthened. The phenomenon has now been ascribed to mice, fruitflies, nematode worms and yeast. These studies, and Gingeras's original reports, came from microarrays — a technology that relies on the tendency of nucleic acids to find their complementary cousins in a solution. Gingeras works for the microarray manufacturer Affymetrix in Santa Clara, California. But not everyone has been persuaded of the extent of pervasive transcription, in part because microarrays are subject to background 'noise'. Even using no RNA, control chips will give off some signals, and results can be a matter of interpretation.
Yes, false positives may account for some of the observations but I think most scientists recognize that the microchips are actually detecting rare transcripts. The question is whether these rare transcripts are biologically significant or whether they are artifacts like most of the alternative splice variants that made all the headlines a few years ago.
If the transcripts are accidental and nonfunctional then the fact that we see this in mice, fruit flies, nematodes, and yeast isn't a surprise. It is not evidence that the transcripts are functional. We would like to see evidence that most of these transcripts are (1) evolutionarily conserved, (2) reproducibly synthesized from a functional promoter, and (3) abundant enough in vivo to make a difference,
John Mattick, the director of the Centre for Molecular Biology and Biotechnology at the University of Queensland in Brisbane, Australia, has no such qualms. He is a long-time advocate of non-coding RNA's importance. The doubters, he says, "keep regressing to the most orthodox explanation [that the long RNAs are junk]. But they can't just sit on their intellectual backsides and tell us to prove it."
John Mattick is one of the most vocal cheerleaders for non-coding RNA. He maintains that huge amounts of it are biologically functional. His statement is a tacit confession that he has no proof of his claims. What in the world is wrong with asking for "proof" (evidence) whether sitting on our backsides or standing?
Is Mattick advocating science by assertion? It certainly seems that way in many of his papers.
Brosius, J. (2005) Waste not, want not – transcript excess in multicellular eukaryotes. Trends in Genetics 21:287-288 [DOI: 10.1016/j.tig.2005.02.014]
One of the issues in the junk DNA wars is the importance of all those RNAs that are detected in sensitive assays. About 90% of the human genome is complementary to RNAs that are made at some time in some tissue or other. Does this pervasive transcription mean that most of the genome is functional or are most of these transcripts just background noise due to accidental transcription?
The demise of the Central Dogma of Molecular Biology is becoming an annual event. Most recently, it was killed by non-coding RNA (ncRNA) (Mattick, 2003; 2004). In previous years the suspects included alternative splicing, reverse transcriptase, introns, junk DNA, epigenetics, RNA viruses, trans-splicing, transposons, prions, epigenetics, and gene rearrangements. (I’m sure I’ve forgotten some.)
What’s going on? The Central Dogma sounds like the backbone of an entire discipline. If it’s really a “dogma” how come it gets refuted on a regular basis? If it’s really so “central” to the field of molecular biology then why hasn’t the field collapsed?
In order to answer these questions we need to understand what the Central Dogma actually means. It was first proposed by Francis Crick in a talk given in 1957 and published in1958 (Crick, 1958). In the original paper he described all possible directions of information flow between DNA, RNA, and protein. Crick concluded that once information was transferred from nucleic acid (DNA or RNA) to protein it could not flow back to nucleic acids. In other words, the final step in the flow of information from nucleic acids to proteins is irreversible.
Fig. 1. Information flow and the sequence hypothesis. These diagrams of potential information flow were used by Crick (1958) to illustrate all possible transfers of information (left) and those that are permitted (right). The sequence hypothesis refers to the idea that information encoded in the sequence of nucleotides specifies the sequence of amino acids in the protein.
Crick restated the Central Dogma of Molecular Biology in a famous paper published in 1970 at a time when the premature slaying of the Central Dogma by reverse transcriptase was being announced (Crick, 1970). According to Crick, the correct, concise version of the Central Dogma is ...
... once (sequential) information has passed into protein it cannot get out again (F.H.C. Crick, 1958)
The central dogma of molecular biology deals with the detailed residue-by-residue transfer of sequential information. It states that such information cannot be transferred from protein to either protein or nucleic acid. (F.H.C. Crick, 1970)
Announcing the (Premature) Death of the Central Dogma
The central dogma of biology holds that genetic information normally flows from DNA to RNA to protein. As a consequence it has been generally assumed that genes generally code for proteins, and that proteins fulfil not only most structural and catalytic but also most regulatory functions, in all cells, from microbes to mammals. However, the latter may not be the case in complex organisms. A number of startling observations about the extent of non-protein coding RNA (ncRNA) transcription in the higher eukaryotes and the range of genetic and epigenetic phenomena that are RNA-directed suggests that the traditional view of genetic regulatory systems in animals and plants may be incorrect.
Mattick, J.S. (2003) Challenging the dogma: the hidden layer of non-protein-coding RNAs in complex organisms. BioEssays 25:930-939. The central dogma, DNA makes RNA makes protein, has long been a staple of biology textbooks.... Technologies based on textbook biology will continue to generate opportunities in bioinformatics. However, more exciting prospects may come from new discoveries that extend or even violate the central dogma. Consider developmental biology. The central dogma says nothing about the differences between the cells in a human body, as each one has the same DNA. However, recent findings have begun to shed light on how these differences arise and are maintained, and the biochemical rules that govern these differences are only being worked out now. The emerging understanding of developmental inheritance follows a series of fundamental discoveries that have led to a realization that there is more to life than the central dogma.
Henikoff, S. (2002) Beyond the central dogma. Bioinformatics 18:223-225. It will take years, perhaps decades, to construct a detailed theory that explains how DNA, RNA and the epigenetic machinery all fit into an interlocking, self- regulating system. But there is no longer any doubt that a new theory is needed to replace the central dogma that has been the foundation of molecular genetics and biotechnology since the 1950s.
The central dogma, as usually stated, is quite simple: DNA makes RNA, RNA makes protein, and proteins do almost all of the work of biology.
Gibbs. W.W. (2003) The unseen genome: gems among the junk. Sci. Am. 289:26-33.
Unfortunately, there’s a second version of the Central Dogma that’s very popular even though it’s historically incorrect. This version is the simplistic DNA → RNA → protein pathway that was published by Jim Watson in the first edition of The Molecular Biology of the Gene (Watson, 1965). Watson’s version differs from Crick’s because Watson describes the two-step (DNA → RNA and RNA → protein) pathway as the Central Dogma. It has long been known that these conflicting versions have caused confusion among students and scientists (Darden and Tabery, 2005; Thieffry, 1998). I argue that as teachers we should teach the correct version, or, at the very least, acknowledge that there are conflicting versions of the Central Dogma of Molecular Biology.
The pathway version of the Central Dogma is the one that continues to get all the attention. It’s the version that is copied by almost all textbooks of biochemistry and molecular biology. For example, the 2004 edition of the Voet & Voet biochemistry textbook says,
In 1958, Crick neatly encapsulated the broad outlines of this process in a flow scheme he called the central dogma of molecular biology: DNA directs its own replication and its transcription to yield RNA, which, in turn, directs its translation to form proteins. (Voet and Voet, 2004)
If the Watson pathway version of the Central Dogma really was the one true version then it would have been discarded or modified long ago. In his original description, Watson drew single arrows from DNA to RNA and from RNA to protein and stated ....
The arrow encircling DNA signifies that it is the template for its self-replication; the arrow between DNA and RNA indicates that all cellular RNA molecules are made on DNA templates. Most importantly, both these latter arrows are unidirectional, that is, RNA sequences are never copied on protein templates; likewise, RNA never acts as a template for DNA.
Fig. 2. Watson’s version of the Central Dogma. This figure is taken from the first edition of The Molecular Biology of the Gene (p. 298).
Watson's statement is clearly untrue, as the discovery of reverse transcriptase demonstrated only a few years after his book was published. Furthermore, there are now dozens of examples of information flow pathways that are more complex than the simple scheme shown in Watson’s 1965 book. (Not to mention the fact that many information flow pathways terminate with functional RNA’s and never produce protein.)
Watson’s version of the Central Dogma is the one scientists most often refer to when they claim that the Central Dogma is dead. The reason it refuses to die is because it is not the correct Central Dogma. The correct version has not been refuted.
Crick was well aware of the difference between his (correct) version and the Watson version. In his original 1958 paper, Crick referred to the standard information flow pathway as the sequence hypothesis. In his 1970 paper he listed several common misunderstandings of the Central Dogma including ....
It is not the same, as is commonly assumed, as the sequence hypothesis, which was clearly distinguished from it in the same article (Crick, 1958). In particular, the sequence hypothesis was a positive statement, saying that the (overall) transfer nucleic acid → protein did exist, whereas the central dogma was a negative statement saying that transfers from protein did not exist.
The Sequence Hypothesis and the Central Dogma in 1957
My own thinking (and that of many of my colleagues) is based on two general principles, which I shall call the Sequence Hypothesis and the Central Dogma. The direct evidence for both of them is negligible, but I have found them to be of great help in getting to grips with these very complex problems. I present them here in the hope that others can make similar use of them. Their speculative nature is emphasized by their names. It is an instructive exercise to attempt to build a useful theory without using them. One generally ends in the wilderness.
The Sequence Hypothesis. This has already been referred to a number of times. In its simplest form it assumes that the specificity of a piece of nucleic acid is expressed solely by the sequence of its bases, and that this sequence is a (simple) code for the amino acid sequence of a particular protein.
This hypothesis appears to be rather widely held. Its virtue is that it unites several remarkable pairs of generalizations: the central biochemical importance of proteins and the dominating role of genes, and in particular of their nucleic acid; the linearity of protein molecules (considered covalently) and the genetic linearity within the functional gene, as shown by the work of Benzer and Pontecorvo; the simplicity of the composition of protein molecules and the simplicity of nucleic acids. Work is actively proceeding in several laboratories, including our own, in an attempt to provide more direct evidence for this hypothesis.
The Central Dogma. This states that once “information” has passed into protein it cannot get out again. In more detail, the transfer of information from nucleic acid to nucleic acid, or from nucleic acid to protein may be possible, but transfer from protein to protein, or from protein to nucleic acid is impossible. Information means here the precise determination of sequence, either of bases in the nucleic acid or of amino acid residues in the protein.
Crick, F.H.C. (1958) On protein synthesis. Symp. Soc. Exp. Biol. XII:138-163 quoted in Judson, H.F. The Eight Day of Creation, Expanded Edition (1979, 1996) p. 332.
So, how do we explain the current state of the Central Dogma? The Watson version is the one presented in almost every textbook, even though it is not the correct version according to Francis Crick. The Watson version has become the favorite whipping boy of any scientist who lays claim to a revolutionary discovery, even though a tiny bit of research would uncover the real meaning of the Central Dogma of Molecular Biology. The Watson version has been repeatedly refuted or shown to be incomplete, and yet it continues to be promoted as the true Central Dogma. This is very strange.
The Crick version is correct—it has never been seriously challenged—but few textbooks refer to it. One exception is Lewin’s GENES VIII (Lewin, 2004) (and earlier editions). Lewin defines the Central Dogma of Molecular Biology as,
The central dogma states that information in nucleic acid can be perpetuated or transferred but the transfer of information into protein is irreversible. (B. Lewin, 2004)
I recommend that all biochemistry and molecular biology teachers adopt this definition—or something very similar—and teach it in their classrooms.
Crick, F.H.C. (1958) On protein synthesis. Symp. Soc. Exp. Biol. XII:138-163. [PDF]
Crick, F. (1970) Central Dogma of Molecular Biology. Nature 227, 561-563. [PDF file]