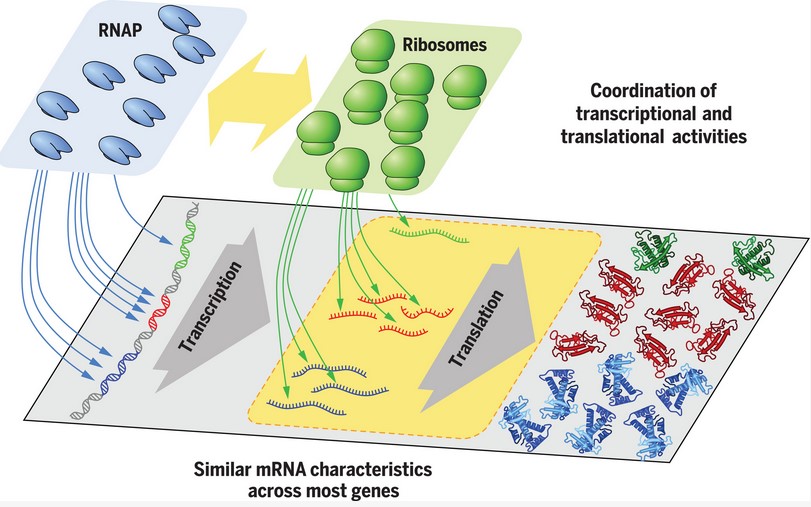
The amount of a given protein in Escherichia coli depends on a number of factors such as the amount of mRNA and the rate of translation. The standard model of regulation is based on decades of study of individual genes and it reveals that the amount of protein is mostly dependent on the amount of mRNA that was translated. This, in turn, indicates that most regulation occurs at the level of transcription initiation.
It's now possible to look simultaneously at the characteristics of large numbers of protein-coding genes to see whether this generality holds. That's what Balakrishan et al. (2022) reported in a Science paper a few years ago. They looked at the characteristics of 1900 protein-coding genes in E. coli to see how protein concentration was regulated.
The expression of genes is regulated at many levels but one of the most important is regulation at the level of transcription. Transcription initiation is controlled by transcription factors that bind to sequences near the promoter and either activate or repress transcription.
A lot of work has been done on transcription regulation in mammals over the past 40 years. The general impression from these detailed studies of individual genes is that regulation usually involves a relatively small number of transcription factors that bind to sequences within 1000 bp or so of the transcription start site.
This model was challenged by the ENCODE studies in 2012. ENCODE researchers claimed to have discovered hundreds of thousands of cis-regulatory elements (CRE's) covering a substantial percentage of the genome. If they are correct, then this means that there are dozens of transcription factors controlling the expression of every gene.
A recent paper on characterizing endogenous retrovirus sequences has attracted some attention because of a press release from Kyoto University that focused on refuting junk DNA. But it turns out that there's no mention of junk DNA in the published paper.
Let's start with a little background. Retroviruses are RNA viruses that go though a stage where their RNA genomes are copied into DNA by reverse transcriptase. The virus may integrate into the host genome and be carried along for many generations producing low levels of virus particles [Retrotransposons/Endogenous Retroviruses]. The integrated copies are called endogenous retroviruses (ERVs).
Our genome contains about 31 different families of ERVS that have integrated over millions of years. Most of the original virus genomes have acquired mutations, including insertions and deletions, and they are no longer active. These sequences account for about 8% of our genome.
There are a million potential transcription regulatory sites in the human genome. How many of these function as true regulatory sites?
One of the important questions about the human genome concerns how gene expression is regulated. The main controversy is over the number of functional regulatory sites and how that relates to abundant junk DNA. Here's how one group addresses the problem by looking at the conservation of regulatory sites in mammals. Sequence conservation is best genomics proxy for identifying functional sites.
Andrews, G., Fan, K., Pratt, H.E., Phalke, N., Zoonomia Consortium, Karlsson, E.K., Lindblad-Toh, K., Gazal, S., Moore, J.E. and Weng, Z. (2023) Mammalian evolution of human cis-regulatory elements and transcription factor binding sites. Science 380:eabn7930. [doi: 10.1126/science.abn7930]
Understanding the regulatory landscape of the human genome is a long-standing objective of modern biology. Using the reference-free alignment across 241 mammalian genomes produced by the Zoonomia Consortium, we charted evolutionary trajectories for 0.92 million human candidate cis-regulatory elements (cCREs) and 15.6 million human transcription factor binding sites (TFBSs). We identified 439,461 cCREs and 2,024,062 TFBSs under evolutionary constraint. Genes near constrained elements perform fundamental cellular processes, whereas genes near primate-specific elements are involved in environmental interaction, including odor perception and immune response. About 20% of TFBSs are transposable element–derived and exhibit intricate patterns of gains and losses during primate evolution whereas sequence variants associated with complex traits are enriched in constrained TFBSs. Our annotations illuminate the regulatory functions of the human genome.
The authors introduce the issue by pointing out two different views of functional regulatory sites. First, there's the ENCODE view, which maps the binding sites of 1600 transcription factors and the associated methylation and histone modification patterns. This analysis creates a database of almost one million candidate cis-regulatory elements (cCREs). Second, there's the evolutionary perspective, which looks at conservation of regulatory sites as the prime indicator of function. Only a fraction of candidate sites are conserved. Does this mean that most of the cCREs are not functional?
Andrews et al. set out to identify all of the cCRE's and transcription factor binding sites (TFBSs) that show evidence of conservation using an alignment of 241 mammalian genomes from the Zoonomia database and a program called phyloP.
They began with more than 920,000 human cCREs from the ENCODE Consortium results. Their results indicate that 47.5% of all CREs are highly conserved because they align to almost all of the 240 non-human mammalian genomes. (I have no idea how the phyloP program calculates "conservation.") That means approximately 439,000 sites that are likely to be genuine regulatory sequences covering 4% of the human genome. If there are 25,000 genes then this means that each gene requires about 17 regulatory sequences.
The next step was to examine 15.6 million TFBSs with a median length of 10 bp covering 5.7% of the human genome. They classified 32.5% of these sequences as highly conserved using the mysterious phyloP program. That means about 5.1 million functional transcription factor binding sites, but later on they reduce this to 2 million covering 0.8% of the genome. This is equivalent to an average of 80 per gene.
I don't believe that the authors have identified functional sites. There is no critical analysis of the results or the methodology and no attempt to rationalize the extraordinary claim that every gene requires so many regulatory sites. About 10,000 genes are regular housekeeping genes, such as those encoding the standard metabolic enzymes, and it's difficult to imagine that those genes require such complex regulation.
John Mattick continues to promote the idea that he is leading a paradigm shift in molecular biology. He believes that he and his colleagues have discovered a vast world of noncoding genes responsible for intricate gene regulation in complex eukaryotes. The latest salvo was fired a few months ago in June 2023.
Mattick, J.S. (2023) A Kuhnian revolution in molecular biology: Most genes in complex organisms express regulatory RNAs. BioEssays:2300080. [doi: 10.1002/bies.202300080]
Thomas Kuhn described the progress of science as comprising occasional paradigm shifts separated by interludes of ‘normal science’. The paradigm that has held sway since the inception of molecular biology is that genes (mainly) encode proteins. In parallel, theoreticians posited that mutation is random, inferred that most of the genome in complex organisms is non-functional, and asserted that somatic information is not communicated to the germline. However, many anomalies appeared, particularly in plants and animals: the strange genetic phenomena of paramutation and transvection; introns; repetitive sequences; a complex epigenome; lack of scaling of (protein-coding) genes and increase in ‘noncoding’ sequences with developmental complexity; genetic loci termed ‘enhancers’ that control spatiotemporal gene expression patterns during development; and a plethora of ‘intergenic’, overlapping, antisense and intronic transcripts. These observations suggest that the original conception of genetic information was deficient and that most genes in complex organisms specify regulatory RNAs, some of which convey intergenerational information.
This paper is promoted by a video in which he explains why there's a Kuhnian revolution under way. This paper differs from most of his others on the same topic because Mattick now seems to have acquired some more knowledge of the mutation load argument and the neutral theory of evolution. Now he's not only attacking the so-called "protein centric" paradigm but also the Modern Synthesis. Apparently, a slew of "anomalies" are casting doubt on several old paradigms.
This is still a paradigm shaft but it's a bit more complicated than his previous versions (see: John Mattick's paradigm shaft). Now his "anomalies" include not only large numbers of noncoding genes but also the C-value paradox, repetitive DNA, introns, enhancers, gene silencing, the g-value enigma, pervasive transcription, transvection, and epigenetics. Also, he now seems to be aware of many of the arguments for junk DNA but not so aware that he can reference any of his critics.1 His challenges to the Modern Synthesis include paramutation which, along with epigenetics, violate the paradigm of the Moden Synthesis because of non-genetic inheritance.
But the heart of his revolution is still the discovery of massive numbers of noncoding genes that only he and a few of his diehard colleague can see.
The genomic programming of developmentally complex organisms was misunderstood for much of the last century. The mammalian genome harbors only ∼20 000 protein-coding genes, similar in number and with largely orthologous functions as those in other animals, including simple nematodes. On the other hand, the extent of non-protein-coding DNA increases with increasing developmental and cognitive complexity, reaching 98.5% in humans. Moreover, high throughput analyses have shown that the majority of the mammalian genome is differentially and dynamically transcribed during development to produce tens if not hundreds of thousands of short and long non-protein-coding RNAs that show highly specific expression patterns and subcellular locations.
The figure is supposed to show that by 2020 junk DNA had been eliminated and almost all of the mammalian genome is devoted to functional DNA—mostly in the form of noncoding genes. There's only one very tiny problem with this picture—it's not supported by any evidence that all those functional noncoding genes exist. This is still a paradigm shaft of the third kind (false paradigm, false overthrow, false data).
1. There are 124 references; Dawkins and ENCODE make the list along with 14 of his own papers. Most of the papers in my list of Required reading for the junk DNA debate are missing. The absence of Palazzo and Gregory (2023) is particularly noteworthy.
Strahl, B. D., and Allis, C. D. (2000) The language of covalent histone modifications. Nature, 403:41-45. [doi: 10.1038/47412]
Histone proteins and the nucleosomes they form with DNA are the fundamental building blocks of eukaryotic chromatin. A diverse array of post-translational modifications that often occur on tail domains of these proteins has been well documented. Although the function of these highly conserved modifications has remained elusive, converging biochemical and genetic evidence suggests functions in several chromatin-based processes. We propose that distinct histone modifications, on one or more tails, act sequentially or in combination to form a ‘histone code’ that is, read by other proteins to bring about distinct downstream events.
They are proposing that the various modifications of histone proteins can be read as a sort of code that's recognized by other factors that bind to nucleosomes and regulation gene expression.
This is an important contribution to our understanding of the relationship between chromatin structure and gene expression. Nobody doubts that transcription is associated with an open form of chromatin that correlates with demethylation of DNA and covalent modifications of histone and nobody doubts that there are proteins that recognize modified histones. However, the key question is what comes first; the binding of transcription factors followed by changes to the DNA and histones, or do the changes to DNA and histones open the chromatin so that transcription factors can bind? These two models are referred to as the histone code model and the recruitment model.
Strahl and Allis did not address this controversy in their original paper; instead, they concentrated on what happens after histones become modified. That's what they mean by "downstream events." Unfortunately, the histone code model has been appropriated by the epigenetics cult and they do not distinguish between cause and effect. For example,
The “histone code” is a hypothesis which states that DNA transcription is largely regulated by post-translational modifications to these histone proteins. Through these mechanisms, a person’s phenotype can change without changing their underlying genetic makeup, controlling gene expression. (Shahid et al. (2022)
The language used by fans of epigenetics strongly implies that it's the modification of DNA and histones that is the primary event in regulating gene expression and not the sequence of DNA. The recruitment model states that regulation is primarily due to the binding of transcription factors to specific DNA sequences that control regulation and then lead to the epiphenomenon of DNA and histone modification.
The unauthorized expropriation of the histone code hypothesis should not be allowed to diminish the contribution of David Allis.
The most important step in the regulation of protein-coding genes in E. coli is the rate of binding of RNA polymerase to the promoter region.
A group of scientists at the University of California at San Diego and their European collaborators looked at the concentrations of proteins and mRNAs of about 2000 genes in E. coli. They catalogued these concentrations under several different growth conditions in order to determine whether the level of protein being expressed from each of these genes correlated with transcription rate, translation rate, mRNA stability or other levels of gene expression.
The paper is very difficult to understand because the authors are primarily interested in developing mathematical formulae to describe their results. They expect you to understand equations like,
even though they don't explain the parameters very well. A lot of important information is in the supplements and I couldn't be bothered to download and read them. I don't think the math is anywhere near as important as the data and the conclusions.